Sickle cell anemia refers to a group of genetic disorders that affect hemoglobin. The disease can be life-threatening, but there are ways to manage the symptoms. Red blood cells contain hemoglobin, which delivers oxygen to the body's tissues. In sickle cell disease, the red blood cells are C-shaped (like a sickle) and sticky. As a result, red blood cells can block blood vessels and are unable to effectively deliver oxygen, resulting in a variety of symptoms.
Genetics Home Handbook , sickle cell disease disproportionately affects “people whose ancestors come from Africa; Mediterranean countries such as Greece, Türkiye and Italy; Arabian Peninsula; India and the Spanish-speaking regions of South America, Central America and parts of the Caribbean."
Types
There are several different types of sickle cell disease. The main types include:
- HbSS
- Sickle cell disease (homozygosity for abnormal HbS): A person inherits two genes, one from each parent, for sickle cell disease. He will have sickle cell anemia, which is the most severe type of sickle cell anemia. - HbSC is a double heterozygous (compound heterozygous) carrier of two abnormal hemoglobins HbS and HbC
: a person inherits the sickle cell gene from one of the parents. A gene is inherited from the other parent that results in a different type of abnormal hemoglobin. This disease is usually less serious. - Sickle cell beta thalassemia HbS
: A person inherits the sickle cell gene from one parent and the gene for beta thalassemia, another type of anemia, from the other parent. - Carriage of the sickle cell trait
: If a person has only one gene, they will not have sickle cell disease, but they can pass the gene on to their children.
Sickle cell disease can affect anyone, but is more common in black people. Sickle cell anemia is more likely in areas where malaria is common. However, patients with sickle cell disease have a lower risk of some severe types of malaria.
Sickle cell anemia
Sickle cell anemia is clinically characterized by symptoms caused, on the one hand, by thrombosis of blood vessels of various organs by sickle-shaped erythrocytes, and on the other, by hemolytic anemia. The severity of anemia depends on the concentration of HbS in the red blood cell: the higher it is, the brighter and more severe the symptoms. In addition, other pathological hemoglobins may be present in erythrocytes: HbF, HbD, HbC, etc. Sometimes sickle cell anemia is combined with thalassemia, and the clinical manifestations may decrease or, on the contrary, increase. In the initial period of the disease, the bone marrow system is predominantly affected: swelling appears, as well as pain due to thrombosis of the vessels feeding the joint and bone. Aseptic necrosis of the femoral head with subsequent infection and osteomyelitis is possible. Hemolytic crises usually develop after infections, have a regenerative or hyporegenerative nature and are the main cause of death in these patients. In rare cases, a sequestration crisis is observed due to the deposition of blood in the spleen and liver, which is expressed by abdominal pain syndrome due to the rapid enlargement of these organs and is accompanied by collapse; in this case, hemolysis may be absent; pulmonary infarctions occur due to impaired microcirculation at the level of the pulmonary vessels.
In the second period, a constant symptom is hemolytic anemia. Bone marrow hyperplasia developing in the tubular bones (active hematopoiesis occurs in them as a compensatory reaction to hemolysis) is accompanied by characteristic skeletal changes: thin limbs, a curved spine, a tower-shaped skull with convexities in the forehead and parietal bone. Hepato- and splenomegaly develop due to the activation of erythropoiesis, as well as secondary hemochromatosis and thrombosis; In some patients, cholelithiasis develops. Hemosiderosis of the heart muscle leads to heart failure, and hemosiderosis of the liver and pancreas leads to cirrhosis of the liver and diabetes mellitus.
Renal vascular thrombosis occurs with hematuria and subsequent renal failure. Neurological symptoms are caused by stroke, cranial nerve palsy, etc. Trophic ulcers on the lower extremities are characteristic. Most patients with severe sickle cell anemia die within 5 years, and those who survive this period enter the third period, which is characterized by signs of mild hemolytic anemia. Their spleen is usually not palpable, since repeated infarctions lead to its shrinkage—autosplenectomy. The liver remains enlarged, unevenly compacted, and frequent infections often take a septic course.
Hematological changes. Hemoglobin concentration decreases (
Heterozygous patients feel practically healthy; anemia and morphological changes in red blood cells are detected in them only under hypoxic conditions (climbing mountains, heavy physical activity, flying on airplanes, etc.). However, a hemolytic crisis can also be fatal in them.
Thus, the clinic of sickle cell anemia is characterized by polysymptoms: jaundice of the skin, hypoxic syndrome, hepatosplenomegaly, skeletal deformation, repeated organ thrombosis; from hematological symptoms: regenerative anemia, sickling of erythrocytes detected by special tests, hyperbilirubinemia due to the free fraction. A person’s belonging to a certain ethnic group gives reason to suspect this disease and begin a targeted examination to confirm or rule out this anemia.
A sickle cell crisis includes the following; thrombotic crisis (vaso-occlusive-infectious); hemolytic crisis, characterized by a sharp increase in hemolysis and accompanied by a catastrophic drop in hematocrit with the simultaneous development of jaundice; aplastic crisis, often provoked by an intercurrent infection causing bone marrow suppression or folic acid deficiency with a life-threatening decrease in hematocrit and reticulocyte count in the peripheral blood; sequestration crisis (an unusual symptom of sickle cell anemia in children), clinically manifested by a sudden painful enlargement of the liver and spleen with concomitant pancytopenia.
Thrombotic sickle cell crisis, observed, of course, more often than hemolytic or aplastic, entails a series of progressive pathological changes, culminating in a clinical condition with intractable pain, prostration and anxiety of the patient. Initially, gnawing pain may occur locally - in the tibia, in the periarticular areas, in the back, abdomen or chest. Paroxysmal pain may persist for only a few hours, but subsequently it returns with greater force and is longer lasting. The described state of impending crisis, apparently, can occur in patients with sickle cell anemia who experience vague discomfort with scant physical examination data (examination) and the presence of symptoms that at first appear to be easily remedied by analgesics and intravenous fluids. Any experienced physician has undoubtedly seen such patients repeatedly seek help over a 48- to 72-hour period before culminating in increasing pain requiring hospitalization. The diagnosis of thrombotic crisis is made on the basis of clinical data (before obtaining laboratory confirmation); The significance of recent reports of increased blood alpha-hydroxybutyrate dehydrogenase activity in such patients has not yet been proven. The increase in hemolytic activity can be judged (if initial data is available) by an increase in the number of reticulocytes in the peripheral blood and a decrease in hematocrit. Follow-up laboratory tests are aimed at identifying precipitating factors, including pneumonia, urinary infection, pharyngitis, acidosis and dehydration.
The mainstays of therapy for vaso-occlusive crisis remain pain relief, rehydration, correction of acidosis, and treatment of infection. Additional oxygen administration is possible, but it has little effect on the oxygen tension in the stagnant microvasculature. Red blood cell transfusion is reserved for patients with significant anemia (hematocrit less than 17-18%), since free transfusion with a hematocrit greater than 30% can increase blood viscosity and increase red blood cell aggregation with increased microvascular occlusion. Continuous additional administration of folic acid is indicated, taking into account the increased production of red blood cells with a shortening of their lifespan. The clinical usefulness of drugs that suppress the formation of sickle cell erythrocytes (urea, sodium cyanate) has not been proven.
Sickle cell anemia - symptoms
If the body's cells do not receive enough oxygen, various symptoms and complications develop. It can happen at any age and symptoms will vary from person to person. Sickle cells are destroyed more easily than healthy red blood cells. This can lead to low levels of red blood cells (RBCs), known as anemia.
Early symptoms may include:
- jaundice, or yellowing of the skin and whites of the eyes
- fatigue
- pain and swelling in the arms and legs
Further symptoms and complications may include:
- episodes of pain
- swelling in the arms and legs
- acute chest syndrome
- vision loss
- enlarged spleen
- leg ulcers
- stroke
- deep vein thrombosis
- damage to the liver, heart or kidneys
- cholelithiasis (in young people)
- infertility (in men)
- pulmonary hypertension, which is high blood pressure in the blood vessels supplying the lungs
- heart failure
- damage to bones and joints that occurs due to low blood supply
- high risk of infections, which may have severe symptoms
- fever
The main symptoms and complications of sickle cell anemia:
- Pain
: During a painful episode, the sickle cells become clogged and prevent blood flow to any part of the body. The pain can range from mild to severe and can last for any period of time. - Infections
: People with sickle cell disease may be at high risk of severe illness from COVID-19, regardless of age. - Chest syndrome
- includes chest pain, cough, fever and difficulty breathing. - Enlarged spleen
: weakness, pale lips, rapid breathing and heart rate, thirst and abdominal pain are noted.
If any of the above symptoms appear, a person needs emergency medical attention.
In infants
Because a person inherits the disease, symptoms of sickle cell disease are observed before birth. A routine blood test at birth will show whether this condition is present. In severe cases, the child may develop aplastic crisis, in which the bone marrow stops producing red blood cells, resulting in severe anemia. The child may have an enlarged spleen. Symptoms include poor appetite and lethargy.
Sickle cell anemia
Terminology
For more information about the composition and biological role of blood, about the essence of anemia, see the material “Anemia.
Blood and bloodlessness." Sickle cell anemia is a severe hereditary disease, also known as drepanocytosis, meniscocytosis, S-hemoglobin disease, Gerrick's syndrome, African hemolytic anemia (hemolytic - Greek lit. "dissolving, decomposing blood"). The modern history of the study of this type of anemia begins, it is believed, in the middle of the 19th century , when an autopsy revealed no spleen in the body of an African slave executed for escaping. Then in 1910 a report appeared, authored by cardiology professor James B. Herrick and his intern Ernest E. Irons. In a blood test of a 20-year-old student who had been treated since 1904 for “anemia, muscular rheumatism and biliousness” (he died of pneumonia in 1916), Irons observed and first described red blood cells of a strange shape, which he defined as “elongated and sickle-shaped.” " The name "sickle cell anemia" was first used as a diagnosis by Verne Mason in 1922 . Since the 1930s, epidemiological studies of this disease began in samples of children of African descent.
It can be assumed that the genetic mutation leading to sickle cell anemia appeared on the African continent - and, oddly enough, became established in the population as a useful trait: carriers of the gene and patients with this form of anemia show relative resistance to malaria. The malarial plasmodium parasitizes red blood cells and destroys them, however, red blood cells, genetically mutilated and carrying a completely different hemoglobin (see below), are “inedible” for plasmodium. And although the anemia of malaria is not sweeter, the mortality rate of swamp fever is still higher: those who had at least some immunity survived.
In an actively migrating humanity, which is becoming increasingly international and gradually interracial, sickle cell anemia is widespread. However, a certain endemicity remains today: in Equatorial Africa, the Mediterranean basin, India, and the Middle East, the frequency of genetic carriage (not to be confused with the incidence in the clinical form) reaches 40%. According to a special study conducted in 2001 in Jamaica, the average life expectancy of patients with homozygous sickle cell anemia is 53 years for men and 58 years for women. About 90% live to be 20 years old, and about half live to be 50 years old.
In other words, the problem is very serious, it is characterized by high medical and social significance and requires effective solutions, the search for which is currently being carried out by leading specialized research centers. The prevalence of hereditary hemoglobinopathies (including sickle cell anemia) in the world is estimated at 3-7%. In the early 2010s, WHO classified sickle cell disease as a global health problem.
The epidemiological situation and mortality rates in third world countries are unknown or not sufficiently reliable.
According to the literature, due to migration processes, the incidence in Russia shows a significant and stable tendency to increase, especially in Moscow and St. Petersburg.
Sickle cell anemia - diagnosis
Newborn screening involves a simple blood test to detect sickle cell disease and carrier status of the sickle cell trait. If tests show that your child has sickle cell disease, doctors will monitor him over time. Tests are also available before birth, starting at 8-10 weeks of pregnancy.
If a person with a family history of sickle cell disease plans to have children, they can see a genetic counselor and undergo genetic testing to find out if their children are likely to have the disease.
Causes of sickle cell anemia
Sickle cell anemia is caused by a mutation in genes that is inherited. In this case, the HBV gene, which is responsible for the production of hemoglobin, is affected. As a result, a protein with an incorrect position of the beta chain is formed in the body of a sick person (glutamic acid in this chain is replaced by valine).
In this case, hemoglobin continues to be produced, but its electrical properties are disrupted. If the body suffers from a lack of oxygen, the protein changes its structure - it crystallizes and stretches into long chains, that is, it is transformed into hemoglobin S (HbS). Red blood cells react to such deformation by changing their shape. They also lengthen, which leads to thinning of their walls and destruction.
Sickle cell anemia is inherited in an autosomal recessive manner. For pathology to make itself felt, the altered gene must be received from both the father and the mother. This is the so-called homozygous form, in which hemoglobin S molecules and no others will be present in the human blood.
If the mutated gene is present in only one of the parents, then inheritance of the pathology will also occur, but the form of polymorphism will be heterozygous. In this case, the child himself will not suffer from anemia, but will become an asymptomatic carrier of the pathological gene. Hemoglobin A and hemoglobin S will circulate in his blood in different proportions. If a person is healthy, then anemia does not manifest itself in any way, since hemoglobin A is enough to cover all the needs of the body. With severe dehydration or hypoxia, anemia will make itself felt. A person who is an asymptomatic carrier of the mutated HBV gene can pass it on to their children by inheritance.
Sickle cell anemia - treatment
Drug treatment
The following medications may reduce the risk of complications:
- Hydroxyurea
(Hydrea): Helps provide oxygen to the body. It is not safe to use during pregnancy. Studies have not proven the benefit of this drug in children younger than 9 months. - L-Glutamine Oral Powder
(Endary): Helps reduce the number of sickle cells and is suitable from age 5. - Voxelomotor
(Oxbrita): Increases healthy hemoglobin levels. Suitable from 12 years of age. - Crizanlizumab-tmca
: Reduces pain by preventing blood cells from attaching to blood vessels. Suitable from 16 years of age.
Prevention of infections
Doctors recommend that adults and children take precautions to reduce the risk of infection. These include:
- regular hand washing
- stay away from reptiles such as turtles as they can carry salmonella
- Get vaccinations against influenza, pneumococcal disease, and meningococcal disease
Your doctor may prescribe penicillin or another antibiotic for children under 5 years of age.
Blood transfusion and stem cell transplantation
People with severe symptoms may require a blood transfusion or stem cell transplant. Blood transfusions may be necessary if the patient has severe anemia, an enlarged spleen, infection, or other complications.
A stem cell transplant using cells from a healthy donor can cure sickle cell disease, but can be risky. In addition, the stem cells must be precisely selected.
How does sickle cell anemia manifest?
In a person, the symptoms of sickle cell anemia will be more pronounced the worse the health. Also, the clinical manifestations of anemia are affected by the patient’s age, living conditions and the presence of concomitant diseases. It is customary to distinguish several groups of symptoms, including:
- Symptoms of anemia caused by accelerated destruction of red blood cells.
- Symptoms of anemia caused by blocked lumens of blood vessels.
- Symptoms of anemia caused by hemolytic crisis.
The disease does not make itself felt in children under 3 months of age. Sometimes it first manifests itself at the age of 6 months.
Symptoms may include:
- Swelling in the area of the hands and feet, their pain.
- Muscular weakness.
- Changing the shape of the limbs.
- Retardation in physical development in terms of the formation of motor skills.
- Dry skin, paleness.
- With massive destruction of red blood cells, the skin becomes yellow, which occurs due to the release of bilirubin from the destroyed red blood cells into the blood.
Until the child reaches the age of 5-6 years, any infections can have a severe course, which is associated with pathological changes in the spleen. Its lumens become clogged with destroyed red blood cells, and the organ is unable to function normally. It is the spleen in the human body that is designed to cleanse the blood of infectious agents; lymphocytes are also formed in it. Therefore, almost any infection can lead to the development of sepsis. In this regard, parents should be especially vigilant and seek medical help for any illness.
The older the child gets, the more intense his symptoms due to chronic oxygen deprivation become, including:
- The child gets tired quickly.
- He gets dizzy regularly.
- Shortness of breath appears.
- Physical, mental and sexual development is delayed.
Women diagnosed with sickle cell disease can have children, but pregnancy comes with a number of complications.
Symptoms of sickle cell anemia in adolescents and adults:
- From time to time, painful sensations arise in different organs.
- Ulcers often form on the skin.
- Vision deteriorates.
- Kidney failure may develop.
- The structure of the bones changes.
- The joints begin to ache and swell greatly.
- Sensitivity of the limbs deteriorates and paresis occurs.
Hemolytic crisis is a dangerous condition for humans. With sickle cell anemia, it can be triggered by dehydration, severe hypothermia, excessive exercise, and mountain sickness.
The following symptoms indicate a developing crisis:
- Reduced hemoglobin levels to critical levels.
- Fainting conditions.
- High body temperature.
- Urine becomes dark in color.
Lifestyle
Many people with sickle cell disease can live full and active lives, especially if they take steps to reduce the effects of the condition. Lifestyle measures that may help include:
- regular medical examinations
- drink 8 to 10 glasses of water per day
- do not overcool or overheat, as this can provoke a crisis
- engage in physical activity, but also get enough rest
- maintaining a healthy diet.
Scientific article on the topic: Gene therapy will help cure sickle cell anemia.
How to detect sickle cell anemia?
Diagnosis of sickle cell anemia begins with questioning the patient’s complaints and collecting an anamnesis. The symptoms that the disease gives are characteristic of many pathologies, so laboratory tests are required.
The main diagnostic methods include:
- Blood sampling for clinical analysis. In this case, a decrease in the level of red blood cells and hemoglobin in the blood will be detected to levels less than 3.5-4.0 * 1012/l and 120 g/l, respectively.
- A biochemical blood test allows you to diagnose increased levels of bilirubin and free iron in the blood.
Additional diagnostic procedures include:
- Performing a blood test using sodium metabisulfite. When interacting with this substance, red blood cells release oxygen, after which their sickle shape can be visualized.
- Treatment of blood with buffer compounds. HbS is poorly soluble in them.
- Performing hemoglobin electrophoresis, which makes it possible to visualize modified red blood cells. This method also allows you to distinguish a heterozygous mutation from a homozygous one.
Other examination techniques that may be required:
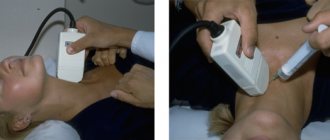
- Ultrasound examination of the liver and spleen, as well as other internal organs to detect pathological changes in them.
- Performing x-rays of the bones of the skeleton and spine.
Anemia - symptoms and treatment
Treatment should first of all be aimed at identifying and eliminating the root cause, so therapy for anemia lasts a long time, often accompanied by several medical specialists.
Indication for treatment
The indication for drug treatment is a decrease in hemoglobin levels to less than 120 g/l in women and below 130 g/l in men.
Basically, treatment involves restoring hemoglobin reserves, increasing the number of red blood cells, and normalizing hematocrit. The main effective method of treating iron deficiency anemia is the use of iron supplements orally or parenterally (intravenously or intramuscularly)
To restore the hemoglobin level in a patient with iron deficiency anemia, the dose of ferrous iron per day (only it is effectively absorbed) must be 100-300 mg, taking into account depleted iron reserves (about 1.5 g).
| Drug name | Components included in the drug | Fe dosage, mg | Dosage form of release | Daily dose, g |
| Conferon | succinic acid | 50 | Pills | 3-4 |
| Heferol | Fumaric acid | 100 | Capsules | 1-2 |
| Hemophere prolongatum | Ferrous sulfate | 105 | Dragee | 1-2 |
| Ferrogradumet | Plastic matrix - gradation | 105 | Pills | 1-2 |
| Aktiferrin | D, L-serine | 113,8 34,8 | Capsules Syrup | 1-2 1 teaspoon per 12 kg body weight |
| Ferroplex | Ascorbic acid | 10 | Dragee | 8-10 |
| Sorbifer Durules | Ferrous sulfate + Ascorbic acid | 100 | Pills | 1-2 |
| Tardiferon | Ferrous sulfate + mucoprotease | 80 | Pills | 1-2 |
| Fenyuls | Ascorbic acid, nicotinamide, B vitamins | 50 | Capsules | |
| Irovit | The same + ascorbic acid, cyanocobalamin, L-lysine | 100 | Capsules | 1-2 |
| Irradian | Ascorbic acid, folic acid, cyanocobalamin, L-cysteine, D-fructose, yeast | 100 | Dragee | 1-2 |
It is more effective to prescribe drugs with a higher content of ferrous iron; they should be taken orally (by mouth) 1-2 times a day. This is the most convenient approach for patients, and therefore their adherence to therapy increases. Many dosage forms of iron include ascorbic and succinic acids, fructose, cysteine, etc., they help the best absorption of iron in the gastrointestinal tract. Iron supplements are best tolerated if taken with food.
Treatment of iron deficiency anemia with parenteral forms of iron supplements
There are clear indications for such therapy:
- with impaired absorption from the intestine (absorption deficiency syndrome, resection of the small intestine, enteritis, etc.);
- exacerbation of chronic diseases of the gastrointestinal tract, such as peptic ulcer of the stomach or duodenum, Crohn's disease, ulcerative colitis;
- poor tolerance to iron-containing drugs when taken orally;
- if you need to quickly replenish iron reserves in the body.
For parenteral administration, iron preparations are used, such as Ferinject (intravenous), Ektofer (intramuscular), Ferbitol (intramuscular), Ferrum Lek (intramuscular, intravenous), Ferkoven (intravenous) [2].
Regarding the treatment of postpartum anemia, there are no clear conclusions about methods. Studies have shown that oral iron does not have a significant benefit over intravenous iron or placebo. However, oral iron does not cause life-threatening allergic reactions like intravenous iron. The clinical value of blood transfusion remains uncertain, in part because of the risks involved. This method can be used to treat postpartum anemia in cases of acute or severe bleeding during childbirth. However, it is not recommended after minor to moderate bleeding in stable patients with mild or asymptomatic anemia. In any case, it is necessary to assess the severity of anemia and consider the use of an iron-rich diet as a method of prevention and adjunct to treatment [7].
Iron deficiency anemia is the most common type of anemia. However, treatment for other types of anemia differs from treatment for IDA. With other types of anemia, to normalize blood counts, it is necessary, for example, to replenish vitamin B12, folic acid, control concomitant chronic diseases and other factors that were the root cause of anemia. To do this, you need a timely visit to a doctor who will diagnose and determine management tactics.
How to improve treatment. Diet
For all types of anemia, the diet should include foods high in iron: pork and beef liver, veal, beef, buckwheat, green apples, pomegranates, mushrooms, cabbage, beans and other legumes, dark chocolate, etc.
You need to eat 4-6 times a day. For normal digestion, food should be at room temperature; too cold or too hot food irritates the gastric mucosa, which prevents the absorption of beneficial elements. The amount of water you drink per day is 30 ml per 1 kg of weight, including tea, juice, soup, etc. Drinking alcohol and smoking are strictly prohibited. Alcohol not only negatively affects the health of the gastrointestinal tract, but also washes out beneficial components in the urine, which is harmful even for healthy people. Smoking increases stomach acidity, which can cause gastritis or ulcers. In this case, the absorption of beneficial microelements will be impaired.
For iron deficiency anemia, it is necessary to include fermented foods (kefir, kvass, sauerkraut, etc.). The acid contained in such products easily comes into contact with iron and prevents the formation of poorly absorbed iron phytates, resulting in improved penetration of iron into enterocytes (cells lining the inner surface of the intestine). The formation of iron phytates is also reduced if plant products are crushed or cooked [10].
Daily iron supplementation in menstruating women is an effective clinical public health strategy for alleviating anemia and iron deficiency, as well as increasing hemoglobin and iron stores. Daily iron supplementation also improves physical performance in women. In addition, there is evidence that iron supplementation reduces fatigue [8].
Daily oral iron and folic acid supplementation of 30–60 mg elemental iron and 400 μg (0.4 mg) folic acid is recommended for pregnant women to prevent anemia, puerperal sepsis, preterm birth, and low birth weight [9].
Contraindications for anemia
For anemia, strict diets are contraindicated; the diet should include foods high in iron, vitamin B12 and folic acid. The development of anemia is also possible while taking certain medications (antitumor drugs, a number of antibacterial, antiprotozoal, antiviral, anti-inflammatory, antirheumatic, antiepileptic and antipsychotic drugs). Therefore, taking this group of drugs for anemia should be under the supervision of a specialist.
Is it possible to treat anemia with folk remedies?
To date, there are no effective folk remedies for treating anemia, because correction of iron deficiency cannot be achieved only by changing diet or using any herbs, decoctions and juices. The reason for this is that the absorption of iron from food is limited (no more than 3-5% from plant foods), and in medications it is contained in higher concentrations.