Pharmacological properties of the drug Leflunomide
a basic antirheumatic drug with antiproliferative, immunosuppressive and anti-inflammatory properties. The active metabolite of leflunomide, A771726, inhibits the enzyme dehydroorotate dehydrogenase and exhibits antiproliferative activity. A771726 in vitro inhibits mitogen-induced proliferation and DNA synthesis of T lymphocytes. The antiproliferative activity of A771726 appears to manifest itself at the level of pyrimidine biosynthesis, since the addition of uridine to the cell culture eliminates the inhibitory effect of the A771726 metabolite. Using radioisotope ligands, it was shown that A771726 selectively binds to dehydroorotate dehydrogenase, which explains its ability to inhibit this enzyme and the proliferation of lymphocytes at the G1 stage. Lymphocyte proliferation is one of the key stages in the development of rheumatoid arthritis. At the same time, A771726 inhibits the expression of interleukin-2 receptors and nuclear antigens Ki-67 and PCNA, which are associated with the cell cycle. The therapeutic effect usually appears after 4–6 weeks and may further increase over 4–6 months. Leflunomide quickly turns into an active metabolite - A771726 (primary metabolism occurs in the intestines and liver). Trace amounts of unchanged leflunomide are found in plasma, urine or feces. The only detectable metabolite is A771726, which is responsible for the main properties of the drug in vivo . When taken orally, absorption is 82–95%. The maximum concentration of A771726 is achieved 1–24 hours after a single dose. Eating does not affect the absorption of the drug. At a dose of 5–25 mg, the pharmacokinetic parameters of A771726 have a linear relationship. At a dose of 20 mg per day, the average plasma concentrations of A771726 at steady state were 35 μg/ml. A771726 quickly binds to albumin. Unbound fraction A771726 - 0.2%. The association with A771726 proteins is more variable and is slightly reduced in patients with rheumatoid arthritis or chronic renal failure. Leflunomide is metabolized to one major (A771726) and several minor metabolites, including 4-trifluoromethylalanine. A771726 is excreted slowly, clearance is 31 ml/h. The half-life is 2 weeks. In patients with chronic renal failure, the half-life is prolonged.
Psoriatic arthritis (PA) is a chronic inflammatory disease of the joints, spine and entheses, usually associated with psoriasis, and belongs to the group of seronegative spondyloarthropathies. Data on the incidence of PA among patients with psoriasis vary widely - from 7 to 30%, which is due to the characteristics of population studies, as well as the lack of uniform diagnostic criteria for a long time.
Clinical picture of PA
Clinically, PA has features of both rheumatoid arthritis (RA) and ankylosing spondylitis (ASA). PA usually begins between the ages of 30 and 55, and men and women are affected with equal frequency. The exception is isolated psoriatic spondyloarthritis, which is twice as common in men. In most patients (75%), psoriasis precedes arthritis by several months to several years. In 10–15% of patients, arthritis develops before psoriasis, and in 11–15% it develops simultaneously with skin lesions [1, 2].
Damage to the spine (spondylitis) brings the VA closer to the ASA: changes are localized in the iliosacral joints, ligamentous apparatus of the spine with the formation of syndesmophytes and paravertebral ossifications. However, unlike ASA in PA, radiological changes in the sacroiliac joints rarely reach the stage of ankylosis and spinal mobility remains quite satisfactory for a long time.
Previously, PA was considered a “favorable” inflammatory joint disease from the point of view of the course and prognosis. However, in recent years it has been established that in PA, as in RA, the articular syndrome tends to progress and develop destructive changes, which leads to significant impairment of the functional capabilities of patients, especially in the case of mutilating (disfiguring) arthritis or ischemic necrosis of large (supporting) joints. In addition, with this disease, an increase in mortality compared to the general population and a decrease in the quality of life of patients was revealed. The standard mortality rate (SMR) among people suffering from PsA is higher than in the general population – 59% in women and 65% in men [3]. Both psoriasis and arthritis have a negative impact on quality of life. Patients experience serious psychological and physical difficulties not only due to extensive skin damage, but also due to the forced limitation of daily activities as a result of arthritis. The main clinical manifestations of psoriasis and PA are presented in Fig. 1 and .
General principles of treatment of PA
An ideal treatment for PA can be considered a drug that has a positive effect on both the skin and arthritis.
According to the recommendations of the British Society of Rheumatology (2005), the treatment of PsA consists of two main areas - standard therapy and the use of biological agents [4]. The goal of PA therapy is to reduce inflammation in the joints, spine and entheses, slow down radiological progression, and improve the quality of life of patients.
Treatment with biological agents is based on parenteral (intravenous or subcutaneous) administration of drugs that block tumor necrosis factor alpha (TNF-alpha). These primarily include monoclonal antibodies to TNF-alpha infliximab (Remicade) and adalimumab (Humira), which quickly and effectively affect the main clinical manifestations of PsA, but their widespread use in clinical practice is still limited by the high cost of treatment. Therefore, the first and mandatory step in the treatment of PA remains the use of basic anti-inflammatory drugs (DMARDs).
Standard treatment of PA includes the use of non-steroidal anti-inflammatory drugs (NSAIDs), mainly traditional ones - diclofenac or indole group drugs, such as indomethacin, in medium therapeutic doses (there is no data on the preference of selective cyclooxygenase-2 inhibitors in PA), and intra-articular administration of glucocorticoids (IAGC) . DMARDs - sulfasalazine, methotrexate, cyclosporine A and leflunomide (LF) - are prescribed alone or in combination.
A meta-analysis of the results of placebo-controlled, cohort and observational studies assessing the effectiveness and tolerability of drugs for standard therapy of PsA showed that their use in the treatment of this disease is limited. NSAIDs and SHAs cause a reduction in arthritis symptoms, but in some patients the positive effect lasts only a short time. Among DMARDs, the most clinically effective are cyclosporine A and parenteral methotrexate in high doses – 1–3 mg/kg every 10 days (evidence level B), as well as sulfasalazine and LF (evidence level A) [5–7].
Studies of leflunomide in PsA
Leflunomide is an anti-inflammatory immunomodulatory drug that was originally developed for the treatment of RA [8]. Since 2005, LF has been recommended for the treatment of PA in Russia. The use of LF in the treatment of PA is explained by the recognition of the key role of T cells in the pathogenesis of this disease.
Activation of T lymphocytes occurs as a result of antigen recognition and with the participation of receptor interactions CD28-CD80/CD86, which causes a complex cascade of immune reactions, resulting in the production of various groups of cytokines (interleukins, interferons, TNF, colony-stimulating factors, chemokines, growth factors). The main role is played by TNF-alpha, a key pro-inflammatory cytokine that regulates inflammation in the skin and synovium through various mechanisms, such as gene expression, migration, differentiation, cell proliferation, and apoptosis. It has been found that in psoriasis, keratocytes receive a signal to hyperproliferate when T lymphocytes release various chemokines and cytokines, including TNF-alpha. At the same time, high levels of TNF-alpha, interleukin-1, and matrix metalloproteinase are found in psoriatic plaques, synovial fluid and synovium [9].
The active metabolite of LF A77 1726 (malononitrilamide) inhibits the proliferation of activated T and B cells, mainly through blocking tyrosine kinase and dehydroorotate dehydrogenase, which are involved in the de novo biosynthesis of pyrimidine nucleotides. Other biological effects of LF include a decrease in the synthesis of matrix metalloproteinase and the production of immunoglobulins
B cells, the ability to reduce the expression of cyclooxygenase-2 by macrophages, inhibit the activation of nuclear factor kB (NF-kB), disrupt the glycosylation of adhesion molecules, inhibiting intercellular contacts and slowing down the migration of cells to the site of inflammation. A77 1726 suppresses the production of TNF-alpha and interleukin-1b, the proliferation of epidermal cells through the induction of the negative cell cycle regulator p53. The mechanisms of action of LF are covered in detail in previous publications and reviews by other authors [10, 11].
The first reports of the use of LF in PA appeared in 2001. By this time, the drug was actively used in RA, and its role in the treatment of PA was unclear. In this regard, studies regarding the effectiveness and safety of LF for PsA were initially of an open observational nature and were carried out on small groups of patients.
Liang C. and Barr W. observed 12 patients with polyarticular (rheumatoid-like) form of PsA and ineffectiveness of previous DMARD therapy (methotrexate, sulfasalazine, auronaphine, azathioprine) for 8 to 24–31 months. LF was prescribed at a dose of 100 mg/day for 3 days (loading dose), then 20 mg/day daily. Two patients continued taking prednisolone 10 mg/day.
The effectiveness of therapy was assessed at the 2nd and 3rd months of treatment and after the end of LF administration based on changes in the following indicators:
- number of painful and swollen joints;
- hand grip force;
- assessment of PA activity by the patient and physician using a visual analogue scale (VAS; 0–100 mm);
- the doctor’s assessment of the effect of treatment on skin psoriasis and arthritis on a scale from 0 to 3 (0 – no effect, 1, 2 and 3 – weak, moderate and noticeable improvement, respectively).
The majority of patients (eight people) noted a decrease in the number of painful and swollen joints, an increase in hand grip strength, a decrease in disease activity according to VAS, with a simultaneous weakening of psoriasis symptoms, and in seven of them, there was no previously reported effect from taking at least three DMARDs. In general, LF was well tolerated by patients. Among the adverse events, the most significant can be considered the following: one patient developed symptoms of sensory neuropathy of the lower extremities; one patient developed uterine bleeding while taking LF and hormone replacement therapy (in both cases, treatment was discontinued). In four patients, due to the appearance of signs of toxicity, the drug was temporarily suspended and resumed at a lower dose (10 mg/day) after their disappearance. The drug was also discontinued due to increased levels of blood transaminases (one patient), development of alopecia (one patient), and diarrhea (two patients). Half of the patients did not experience any adverse events [12, 13].
Scarpa R. et al. (2001) conducted a pilot study of the effectiveness of LF at a standard dose in six patients with polyarticular form of PA for 3 months. During treatment, there was a significant decrease in the number of painful and swollen joints, a decrease in ESR and the level of C-reactive protein (CRP), but the area of skin lesions remained the same. There were no undesirable effects on liver function [14].
Reich K et al. (2002) reported the successful use of LF in a patient with a long history (more than 44 years) of widespread psoriasis and osteolytic polyarticular form of PsA (more than 15 years) with no effect from previous treatment with DMARDs (cyclosporine A 2 mg/kg for 6 months, methotrexate 7.5 mg/week for 9 years and sulfasalazine 2 g/day for 2 months). LF at a dose of 10 mg/day was used as part of a combination regimen that also included prednisolone 10 mg/day and sulfasalazine 2 g/day, which led to a rapid (after 3 weeks) reduction in the number of painful, swollen joints and the area of psoriatic skin lesions without deterioration of liver function and kidneys [10].
Cuchacovich M. and Soto L. (2002) showed the ability of LF to delay radiological progression in PA and cause bone remodeling. This seems especially important, since it has been noted that in PA, none of the known DMARDs is able to prevent the destruction of articular surfaces [15].
The most significant is considered to be an international multicenter placebo-controlled randomized study assessing the effectiveness and safety of LF in the treatment of PsA and psoriasis, conducted in 2004 (Treatment of Psoriatic Arthritis Study - TOPAS). It included 190 people with active PsA and cutaneous psoriasis. The activity of the process was determined based on the number of painful and swollen joints, as well as the area of psoriatic skin lesions (more than 3 painful and swollen joints, more than 3%, respectively) [16, 17].
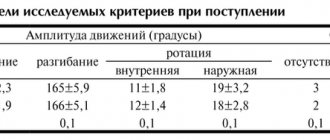
The study results were analyzed in 186 people (91 in the placebo group, 95 in the LF group). Most patients had previous failure of one, and a few patients had, four DMARDs, including methotrexate. At the same time, almost half of the patients included in the study had never taken DMARDs. LF was prescribed at a loading dose of 100 mg/day for 3 days, then 20 mg/day for up to 24 weeks. All patients were comparable in terms of gender, age, race, and disease duration. The frequency of individual clinical variants of PA was the same in both groups, except for mutilating PA, which was more common in the placebo group (see table).
The primary assessment of treatment efficacy was carried out after 24 weeks based on PsARC criteria. Secondary - in the same time frame based on ACR 20 criteria, determination of the dynamics of HAQ (quality of life index) and HAQ DI (dermatological quality of life index), PASI index (change in target plaque) and individual parameters PsARC and ACR [1, 18].
The original PsARC criteria (Psoriatic Arthritis Response Criteria) were proposed by Clegg D. et al. (1996) in a study comparing the effectiveness of sulfasalazine and placebo in PsA. These criteria include a count of painful and swollen joints, a general assessment of PA activity by the doctor and the patient according to
5-point Likert scale [2, 6, 13]:
- a score of painful joints out of 76 on a 4-point scale (from 0 to 3; maximum – 228 points);
- a score of swollen joints out of 74 on a 4-point scale (from 0 to 3; maximum – 222 points);
- general assessment of PA activity by a doctor on a 5-point Likert scale: “excellent”, “good”, “satisfactory”, “bad”, “very bad”;
- general assessment of PA activity by patients on a 5-point Likert scale – “excellent”, “good”, “satisfactory”, “bad”, “very bad”.
ACR criteria modified for PA:
- number of painful joints out of 76;
- number of swollen joints out of 74;
- general assessment of PA activity by a doctor according to VAS (mm);
- general assessment of PA activity by the patient according to VAS (mm);
- assessment of the intensity of joint pain over the last week by the patient according to VAS (mm);
- HAQ (Health Assessment Questionnaire);
- CRP/ESR levels.
The TOPAS study used ACR 20 criteria: a 20% reduction in tender and swollen joints and any 3 of the last 5 ACR scores.
Among patients receiving LF, after 24 weeks, according to PsARC criteria, significantly more patients responded to therapy than in the placebo group: 59 (56/95) versus 30% (27/91).
According to ACR 20 criteria, after 24 weeks, 36.3% (29/80) of patients in the LF group responded to treatment, and 20% (16/80) in the placebo group. In the LF group, compared with the placebo group, there was a significant improvement in other indicators of secondary efficacy assessment (ACR and PsARC components): the number of swollen joints (-4.4 ± 8.6 versus -2.7 ± 9.7), the number of swollen joints joints (-6.8 ± 16.8 vs. -4.2 ± 13.6), number of painful joints (-5.6 ± 10.9 vs. -3.0 ± 12.3), number of painful joints (-9 .1 ± 21.0 versus -4.6 ± 19.6). Also, in patients taking LF, a significant improvement in quality of life indicators was recorded: the change in HAQ score was -0.19 ± 0.51, in the placebo group - -0.05 ± 0.46. The number of patients who noted an improvement in assessing disease activity according to VAS was 52.6% in the LF group and 34.1% in the placebo group.
Treatment with LF led to a significant reduction in the severity and activity of psoriasis: the change in the PASI index in the main group compared to the start of therapy was -2.1 ± 5.9, and when using placebo - only -0.6 ± 6.1 (p = 0.003 ). The number of patients with a decrease in PASI index by 50 and 75% during treatment with LF was 30.4 and 17.4%, and in the placebo group – 18.9 and 7.8%, respectively.
In general, LF was satisfactorily tolerated by patients. Adverse events potentially related to the drug were reported in 61 (63.5%) patients in the LF group and in 37 (40.2%) in the placebo group. There were no cases of respiratory infections or death of patients. Serious adverse events were observed in 13.5% of patients in the LF group and in 5.4% in the placebo group. The most common adverse reactions were increased ALT and/or AST levels and neutropenia. Diarrhea, headache, and nausea were less common.
A study conducted at the Institute of Rheumatology of the Russian Academy of Medical Sciences was devoted to assessing the effectiveness and tolerability of LF in PsA. The results of a preliminary analysis of data from 63 patients were published [19].
The study included patients with active PA, which was defined by the presence of at least five painful and five swollen joints. The majority of patients, namely 85%, had previously taken one or more DMARDs (sulfasalazine, cyclosporine A, methotrexate). Patients received LF according to the standard regimen: loading dose of 100 mg/day for 3 days, then 20 mg/day. The duration of treatment was 24 weeks, the effectiveness and tolerability of therapy was assessed every month. The number of painful and swollen joints was determined, PA activity was assessed by the patient and doctor using VAS, pain intensity was assessed by the patient using VAS, PA activity and psoriasis were assessed by patient and doctor on the Likert scale, the PASI index was determined, and patients independently filled out the HAQ questionnaire. There was a significant decrease in almost all of these parameters after the first month of treatment. Indicators of pain severity and assessment of PA activity in patients improved starting from the 2nd and 3rd months of treatment and subsequently did not change significantly until the end of the study. By the time of completion of treatment, 58% (36/62) of patients responded to LF therapy according to PsARC criteria, and 55% (34/62) according to ACR criteria.
The study was the first to analyze the response to LF therapy in patients with different duration of the disease: less than 3 years (10 patients), from 3 to 10 years (29), more than 10 years (24). It turned out that in patients with a short history of PA, positive dynamics were noted only in relation to the number of swollen joints and HAQ score; in the other two groups, after 6 months, a significant improvement in almost all studied parameters was observed. The number of patients who responded to treatment with LF according to the PsARC and ACR criteria was the same in these groups and did not depend on the duration of PA.
The level of CRP (mg/l) significantly decreased by the third month of LF use. This trend continued by the 6th month of therapy. The ESR indicator did not change significantly throughout the observation period, despite the improvement in all main indicators of clinical activity of PA. This fact confirms the existing opinion about the dissociation of clinical and laboratory activity in this disease, as well as the lack of methods for assessing the latter adequate for PA.
Interesting data have been obtained regarding the effect of LF on the severity of psoriasis. In contrast to the TOPAS study, analysis of PASI dynamics did not reveal a positive effect of LF on the severity and prevalence of psoriasis; moreover, an exacerbation of the skin process was registered in 14 (22%) patients.
Quality of life was assessed based on changes in the total HAQ score after 3 and 6 months from the start of therapy. Thirty (48%) patients experienced a 0.5 decrease in total HAQ score during treatment, which is considered clinically significant.
Tolerability of LF therapy was generally satisfactory. The frequency and severity of side effects did not differ from those described by other authors. Adverse reactions as a reason for premature discontinuation of LF were registered in 13 (21%) patients. These included allergic dermatitis, severe exacerbation of psoriasis, accompanied by ulceration of plaques and the addition of a secondary infection, a more than 2-fold increase in the level of liver enzymes, furunculosis, increased hair loss, and ossalgia.
The results of controlled, uncontrolled and observational studies presented above indicate that LF helps to reduce the symptoms of PsA and psoriasis, improves the quality of life of patients, including those cases where previous use of DMARDs was ineffective. The positive effect on both the manifestations of inflammation in the joints and on the skin allows LF to occupy a worthy place in the basic anti-inflammatory therapy of PA.
Contraindications to the use of the drug Leflunomide
Hypersensitivity to leflunomide, liver failure, severe immunodeficiency (including AIDS), bone marrow hematopoiesis disorders (anemia, leukopenia or thrombocytopenia not associated with rheumatoid arthritis), severe infections, moderate or severe renal failure, severe hypoproteinemia (including nephrotic syndrome), pregnancy and lactation, women of reproductive age (not using contraceptives during treatment with leflunomide and while the plasma level of the active metabolite is above 0.02 mg/l), age under 18 years.
Side effects of the drug Leflunomide
From the cardiovascular system: increased blood pressure. From the gastrointestinal tract: diarrhea, nausea, vomiting, anorexia, erosive and ulcerative lesions of the oral mucosa (aphthous stomatitis, lip ulceration), abdominal pain, increased activity of liver transaminases, especially ALT, less often γ-glutamine transferase, alkaline phosphatase, hyperbilirubinemia, hepatitis, cholestastatic jaundice, liver failure, acute liver necrosis. From the musculoskeletal system: tenosynovitis; ligament rupture. From the skin: increased hair loss, eczema, dry skin. Allergic reactions: rash (including maculopapular rash), skin itching; urticaria, erythema multiforme, malignant exudative erythema (Stevens-Johnson syndrome), toxic epidermal necrolysis (Lyell's syndrome); anaphylactic/anaphylactoid reactions. From the hematopoietic organs: leukopenia, anemia, slight thrombocytopenia (platelets less than 100,000/μl), eosinophilia, pancytopenia; less than 0.01% - agranulocytosis. Laboratory indicators: slight hyperlipidemia, hypouricemia, increased activity of LDH and CPK, slight hypophosphatemia. Other: reversible decrease in sperm concentration, total sperm count and motility; less than 0.01% - development of severe infections and sepsis.
Special instructions for the use of Leflunomide
The drug should be prescribed after a thorough medical examination by specialists who have the necessary experience in the treatment of rheumatoid arthritis. The dosage regimen for mild chronic renal failure has not been determined. Before starting treatment, it is necessary to remember about the possible increase in the number of side effects in patients who have previously received basic drugs for the treatment of rheumatoid arthritis that have hepato- and hematotoxic effects. The active metabolite of leflunomide, A771726, is characterized by a long half-life of 1–4 weeks. Serious adverse effects (eg, hepatotoxicity, hematotoxicity, or allergic reactions) may occur even if leflunomide treatment is discontinued. Therefore, if such cases of toxicity occur or when switching to taking another basic drug (for example, methotrexate) after treatment with leflunomide, it is necessary to carry out a “washing” procedure: cholestyramine - 8 g 3 times a day for 11 days; alternatively, 50 g of activated carbon, crushed into powder, 4 times a day for 11 days. Rare cases of severe liver damage, in some cases fatal, have been reported during treatment with leflunomide, most of which were observed in the first 6 months of treatment. Although the causal relationship of these adverse events to leflunomide has not been established, and in most cases there were multiple risk factors, recommendations for monitoring patients during treatment must be carefully followed. It is necessary to monitor ALT activity before starting therapy, and then every month during the first 6 months of treatment, followed by monitoring once every 2–3 months. Recommendations for adjusting the dosage regimen or discontinuing the drug depending on the severity and persistence of the increase in ALT levels: if the upper limit of the ALT level is confirmed to be exceeded by 2–3 times, reduce the dose from 20 to 10 mg/day (it is possible to continue taking leflunomide subject to careful monitoring this indicator); If a 2- or 3-fold increase in the upper limit of normal for ALT persists, or if there is a rise in the level of ALT that exceeds the upper limit of normal by more than 3 times, leflunomide should be discontinued and a washout procedure initiated. During the treatment period, it is recommended to refrain from taking ethanol (increased risk of hepatotoxicity). A complete clinical blood test, including determination of the leukocyte formula and platelet count, must be performed before starting treatment, as well as every 2 weeks during the first 6 months of treatment and every 8 weeks after completion of treatment. In patients with pre-existing anemia, leukopenia and/or thrombocytopenia, as well as in patients with impaired bone marrow function or at risk of developing such disorders, the risk of hematological disorders increases. If this type of phenomenon occurs, a “washing” procedure should be used to reduce the level of A771726 in the blood plasma. If severe hematotoxicity develops, it is necessary to stop taking leflunomide and any other concomitant drug that inhibits bone marrow hematopoiesis and begin a washout procedure. If ulcerative stomatitis develops, leflunomide should be discontinued. Very rare cases of Stevens-Johnson syndrome or toxic epidermal necrolysis have been reported in patients receiving leflunomide. In case of skin and/or mucous reactions, it is necessary to stop taking leflunomide and any other drug associated with it and immediately begin the “washing out” procedure (in such cases it is essential); re-prescription is contraindicated. Drugs like leflunomide, which have immunosuppressive properties, increase the risk of developing various infections. Infectious diseases that arise are usually severe and require early and intensive treatment. If a severe infectious disease occurs, it is necessary to interrupt treatment with leflunomide and begin a washout procedure. It is necessary to carefully monitor patients with severe tuberculin reactivity due to the risk of reactivation of tuberculosis. Blood pressure levels should be monitored before starting treatment with leflunomide and periodically after starting it. There are no experimental data on the risk of fetotoxicity when using leflunomide in men. To minimize the possible risk, men planning offspring should stop taking leflunomide and use cholestyramine 8 g 3 times a day for 11 days or 50 g of powdered activated carbon 4 times a day for 11 days. Men receiving treatment with leflunomide should be warned about the possible fetotoxic effects of the drug and the need to use adequate contraception. It is necessary to ensure that there is no pregnancy before starting treatment with leflunomide. If you suspect pregnancy, you must immediately inform your doctor. The doctor should discuss with the patient the possible risks to which this pregnancy is exposed. A rapid decrease in the level of the active metabolite in the blood using a drug “washout” procedure if pregnancy is suspected will help reduce the risk to the fetus. If a woman taking leflunomide wishes to become pregnant, it is recommended that one of the following procedures be followed to ensure that the fetus is not exposed to toxic concentrations of A771726 (control concentration below 0.02 mg/L). Waiting period: It is accepted that the plasma concentration of A771726 may be above 0.02 mg/l for an extended period. It is expected that concentrations may fall below 0.02 mg/L 2 years after discontinuation of leflunomide treatment. After this, it is necessary to measure the concentration of A771726 in the blood plasma after at least 14 days. If both measurements are below 0.02 mg/l, there is no risk of teratogenicity. Next comes the “laundering” procedure. Regardless of the laundering procedure chosen, it must be verified by two separate tests at least 14 days apart. Fertilization is possible 1.5 months after the concentration of the active metabolite is below 0.02 mg/l. It is necessary to refrain from conceiving for 2 years after stopping treatment with leflunomide. If a waiting period of approximately 2 years with reliable contraception seems unreasonable, a “washing out” procedure can be performed for preventive purposes. The contraceptive activity of oral contraceptives may be reduced as a result of the “washing” procedure with cholestyramine or activated carbon. It is recommended to use alternative methods of contraception. The experiment showed that leflunomide and its metabolites pass into breast milk.
Buy Leflunomide Canon film-coated tablets 20 mg No. 30 in pharmacies
Instructions for use
Leflunomide tab p.o 20 mg No. 30
Dosage forms tablets 10 mg tablets 20 mg Synonyms Arava Leflide Leflunomide Elafra Group Anti-inflammatory drugs of different groups International nonproprietary name Leflunomide Composition Active substance: leflunomide 10 mg. Manufacturers Canonpharma Production (Russia) Pharmacological action Pharmacodynamics Leflunomide belongs to the class of basic antirheumatic drugs and has antiproliferative, immunomodulatory, immunosuppressive and anti-inflammatory properties. The active metabolite of leflunomide A771726 inhibits the enzyme dehydroorotate dehydrogenase and has antiproliferative activity. A771726 in vitro inhibits mitogen-induced proliferation and deoxyribonucleic acid (DNA) synthesis of T lymphocytes. The antiproliferative activity of A771726 appears to manifest itself at the level of pyrimidine biosynthesis, since the addition of uridine to the cell culture eliminates the inhibitory effect of the A771726 metabolite. Using radioisotope ligands, it was shown that A771726 selectively binds to the enzyme dehydroorotate dehydrogenase, which explains its ability to inhibit this enzyme and the proliferation of lymphocytes at the G1 stage. Lymphocyte proliferation is one of the key stages in the development of rheumatoid arthritis. At the same time, A771726 inhibits the expression of receptors for interleukin-2 (CB-25) and nuclear antigens Ki-67 and PCNA, associated with the cell cycle. The therapeutic effects of leflunomide have been demonstrated in several experimental models of autoimmune diseases, including rheumatoid arthritis. Leflunomide reduces symptoms and slows the progression of joint damage in active rheumatoid arthritis. The therapeutic effect usually appears after 4 - 6 weeks and can further increase over 4 - 6 months. Pharmacokinetics Leflunomide is rapidly converted to its active metabolite A771726 (primary metabolism in the intestinal wall and liver). Only trace amounts of unchanged leflunomide were observed in plasma, urine or feces. The only detectable metabolite is A771726, which is responsible for the main properties of the drug in vivo. When taken orally, 82 to 95% of the drug is absorbed. Maximum plasma concentrations of A771726 are determined from 1 to 24 hours after a single dose. Leflunomide may be taken with food. Due to the very long half-life (T1/2) of A771726 (about 2 weeks), a loading dose of 100 mg per day was used for 3 days. This made it possible to quickly achieve an equilibrium state of the plasma concentration of A771726. Without a loading dose, 2 months of drug administration would be required to achieve steady-state concentrations. In multiple dose studies, the pharmacokinetic parameters of A771726 were dose dependent over the dose range of 5 to 25 mg. In these studies, the clinical effect was closely related to the plasma concentration of A771726 and the daily dose of leflunomide. At a dose of 20 mg per day, the average plasma concentrations of A771726 at steady state were 35 μg/ml. In plasma, A771726 rapidly binds to albumin. The unbound fraction of A771726 is approximately 0.62%. Binding of A771726 is more variable and is slightly reduced in patients with rheumatoid arthritis or chronic renal failure. Leflunomide is metabolized to one major (A771726) and several minor metabolites, including 4-trifluoromethylalanine. The biotransformation of leflunomide to A771726 and the subsequent metabolism of A771726 itself is controlled by several enzymes and occurs in microsomal and other cellular fractions. Interaction studies with cimetidine (a non-specific cytochrome P450 inhibitor) and rifampicin (a non-specific cytochrome P450 inducer) showed that in vivo CYP enzymes are involved in the metabolism of leflunomide only to a small extent. The elimination of A771726 from the body is slow and is characterized by a clearance of 31 ml/hour. Leflunomide is excreted in feces (probably due to biliary excretion) and urine. (T1/2) is about 2 weeks. The pharmacokinetics of A771726 in patients on chronic ambulatory peritoneal dialysis (CAPD) are similar to those in healthy volunteers. A more rapid elimination of A771726 is observed in patients on hemodialysis, which is not due to the extraction of the drug into the dialysate, but to its displacement from the protein. Although the clearance of A771726 increases approximately 2-fold, the final clearance (T1/2) is similar to that in healthy individuals, since the volume of distribution simultaneously increases. There are no data on the pharmacokinetics of the drug in patients with liver failure. Pharmacokinetic characteristics have not been studied in patients under 18 years of age. In elderly patients (65 years and older), pharmacokinetic data approximately correspond to the average age group. effects From the cardiovascular system. Often: moderate increase in blood pressure; rarely: marked increase in blood pressure; frequency unknown: angina pectoris, migraine, palpitations, tachycardia, varicose veins, vasculitis, vasodilation. From the gastrointestinal tract. Common: diarrhea, nausea, vomiting, diseases of the oral mucosa (for example, aphthous stomatitis, lip ulceration), abdominal pain; uncommon: disturbance of taste; very rarely: pancreatitis; frequency unknown: gingivitis, oral candidiasis, esophagitis, gastritis, gastroenteritis, dyspepsia, colitis, constipation, flatulence, melena. From the hepato-biliary system. Often: increased activity of “liver” transaminases (alanine aminotransferase (ALT), gamma-glutamyl transpeptidase (GGT), alkaline phosphatase (ALP), hyperbilirubinemia; rarely: hepatitis, cholestasis, jaundice, cholelithiasis; very rarely: severe liver damage (including liver failure, acute liver necrosis), which can be fatal. On the respiratory system. Common: upper respiratory tract respiratory infections, cough; rare: interstitial pulmonary process (including interstitial pneumonia and pulmonary fibrosis) with possible death; frequency unknown: asthma, shortness of breath, nosebleeds Metabolic and nutritional disorders Common: increased creatine phosphokinase (CPK) Uncommon: hypokalemia, hyperlipidemia, hypophosphatemia Rare: increased lactate dehydrogenase levels Frequency unknown: hypouricemia, diabetes mellitus, hyperthyroidism, peripheral edema Nervous system disorders Common : headache, dizziness, paresthesia; uncommon: anxiety; very rare: peripheral neuropathy; frequency unknown: anxiety, depression, dry oral mucosa, sleep disturbances, neuralgia, neuritis, increased sweating. From the musculoskeletal system. Common: tenosynovitis; uncommon: tendon rupture; frequency unknown: arthralgia, synovitis, muscle spasms, arthrosis, bone necrosis, bursitis, myalgia. From the skin and subcutaneous tissue. Common: increased hair loss, alopecia, eczema, skin rash (including maculopapular), itching, dry skin; uncommon: urticaria; very rare: toxic epidermal necrolysis (Lyell's syndrome), erythema multiforme, Stevens-Johnson syndrome; frequency unknown: acne, contact dermatitis, fungal dermatitis, hair color change, herpes simplex, herpes zoster, nail lesions, skin pigmentation disorders, skin ulceration. From the immune system. Often: allergic reactions; very rare: serious anaphylactic/anaphylactoid reactions, angioedema. Infections and infestations. Rare: severe infections (including opportunistic infections and sepsis), which can be fatal. The risk of infectious diseases, in particular rhinitis, bronchitis and pneumonia, is increased. Hematopoietic system and lymphatic system. Common: leukopenia (leukocytes > 2000/µl); uncommon: anemia, mild thrombocytopenia (platelets > 100,000/µl); rarely: pancytopenia (apparently due to antiproliferative effects), eosinophilia, leukopenia (leukocytes <2000/μl); very rare: agranulocytosis. Recent, concomitant, or subsequent use of potentially myelotoxic agents may be associated with a greater risk of hematologic effects. From the reproductive system. Not known: slight decrease in sperm concentration, total sperm count and motility. From the kidneys and urinary system. Frequency unknown: urinary tract infections, renal failure, albuminuria, cystitis, dysuria, hematuria, prostate damage, frequent urination, vaginal candidiasis. From the senses. Frequency unknown: blurred vision, cataracts, conjunctivitis, taste disturbances. Are common. Common: anorexia, weight loss (usually minor), asthenia; Frequency unknown: fever, weakness. Other. The risk of malignant diseases, especially lymphoproliferative diseases, increases with the use of certain immunosuppressive drugs. Indications for use As a basic treatment for adult patients with active rheumatoid arthritis in order to reduce symptoms of the disease and delay the development of structural damage to the joints; Active form of psoriatic arthritis. Contraindications Hypersensitivity to leflunomide or any other component of the drug; liver dysfunction; severe immunodeficiency (including acquired immunodeficiency syndrome); significant disorders of bone marrow hematopoiesis or severe anemia, leukopenia or thrombocytopenia due to causes other than rheumatoid arthritis; severe, uncontrolled infections; moderate or severe renal failure (creatinine clearance less than 60 ml/min, due to little experience with clinical observation); severe hypoproteinemia (for example, with nephrotic syndrome); in pregnant women or women of childbearing potential who are not using reliable contraception, during treatment with leflunomide and as long as the plasma level of the active metabolite remains above 0.02 mg/l. Pregnancy should be excluded before starting treatment with leflunomide; during breastfeeding (see section “Use during pregnancy and breastfeeding”); children's age (under 18 years), due to the lack of data on effectiveness and safety. Men receiving treatment with leflunomide should be warned about the possible fetotoxic effect of the drug (associated with its possible effect on the father's sperm) and the need to use reliable contraception. Route of administration and dosage Treatment with leflunomide should be initiated under the supervision of a physician experienced in the treatment of rheumatoid arthritis and psoriatic arthritis. The tablets must be taken regardless of food intake, swallowed whole with a sufficient amount of liquid. Treatment with leflunomide begins with an oral loading dose of 100 mg once daily for 3 days. As a maintenance dose for rheumatoid arthritis, it is recommended to take 10 mg to 20 mg of leflunomide once a day, for psoriatic arthritis - 20 mg 1 time a day. The therapeutic effect usually appears after 4 - 6 weeks and can further increase up to 4 - 6 months. No dose adjustment is required for patients over 65 years of age. Overdose Symptoms. There have been reports of chronic overdose in patients receiving leflunomide at doses up to five times the recommended daily dose, as well as reports of acute overdose in adults and children. In most cases of overdose, no adverse reactions were reported. The adverse reactions that occurred were comparable to the safety profile of leflunomide. The most commonly observed adverse reactions were diarrhea, abdominal pain, leukopenia, anemia and increased liver function tests. Treatment. In case of overdose or toxicity, it is recommended to take cholestyramine or activated carbon to speed up the cleansing of the body. Cholestyramine, taken orally 8 g 3 times a day for 24 hours, reduces plasma A771726 levels by approximately 40% after 24 hours, and by 49-65% after 48 hours. Administration of activated carbon (in the form of a suspension) orally or through a gastric tube (50 g every 6 hours during the day) reduces the concentration of the active metabolite A771726 in plasma by 37% after 24 hours, and by 48% after 48 hours. These procedures can be repeated according to clinical indications. Studies with hemodialysis and CAPD indicate that A771726, the major metabolite of leflunomide, is not excreted by dialysis. Interaction Increased side effects may occur in the case of recent or concomitant use of hepatotoxic (including alcohol) or hematotoxic and immunosuppressive drugs, or when these drugs are started after treatment with leflunomide without a washout procedure. In patients with rheumatoid arthritis, no pharmacokinetic interaction was found between leflunomide (10 - 20 mg per day) and methotrexate (10 - 25 mg per week). In patients taking leflunomide, concomitant administration of cholestyramine or activated charcoal is not recommended as this will result in a rapid and significant decrease in plasma concentrations of A771726. This is believed to be due to impaired recycling of A771726 in the liver and small intestine and/or impaired gastrointestinal dialysis. If the patient is already taking non-steroidal anti-inflammatory drugs (NSAIDs) and/or corticosteroids, they can be continued after starting treatment with leflunomide. The enzymes involved in the metabolism of leflunomide and its metabolites are not precisely known. An in vivo study of its interaction with cimetidine (a non-specific cytochrome P450 inhibitor) showed no significant interaction. Following concomitant administration of a single dose of leflunomide to subjects receiving multiple doses of rifampicin (a nonspecific cytochrome P450 inducer), peak levels of A771726 increased by approximately 40%, whereas the area under the concentration-time curve did not change significantly. The mechanism of this effect is not clear. In vitro studies have shown that A771726 inhibits the activity of cytochrome P4502C9 (CYP2C9). In clinical studies, no problems were observed with the concomitant use of leflunomide and NSAIDs metabolized by CYP2C9. Leflunomide should be used with extreme caution with other non-NSAID drugs metabolized by CYP2C9, such as phenytoin, warfarin and tolbutamide. An increase in prothrombin time has been reported with the simultaneous use of leflunomide and warfarin. In a study in which leflunomide was given to healthy female volunteers concomitantly with triphasic oral contraceptives containing 30 mcg ethinyl estradiol, no decrease in the contraceptive effect of the contraceptives was observed, and the pharmacokinetics of A771726 were well within the prescribed range. There is currently no information regarding the concomitant use of leflunomide with antimalarials used in rheumatology (for example, chloroquine and hydroxychloroquine), gold preparations (intramuscular or oral), D-penicillamine, azathioprine and other immunosuppressive drugs (except methotrexate). The risk associated with combination therapy is unknown, especially during long-term treatment. Since this type of therapy can lead to the development of additive or even synergistic toxicity (for example, hepato- or hematotoxicity), combinations of this drug with other basic drugs (for example, methotrexate) are undesirable. Recent concomitant or subsequent use of potentially myelotoxic agents may be associated with a greater risk of hematologic effects. Immunosuppressants increase the risk of developing infections, as well as malignancies, especially lymphoproliferative diseases. Vaccination. There are no clinical data regarding the effectiveness and safety of vaccination in the setting of leflunomide treatment. However, vaccination with live vaccines is not recommended. When planning vaccination with a live vaccine, the long T1/2 of leflunomide after discontinuation of the drug should be taken into account. Special instructions Use during pregnancy and breastfeeding. Leflunomide should not be used by pregnant women or women of childbearing potential who are not using reliable contraception during treatment with leflunomide and for a certain period after this treatment. Before starting treatment with the drug, you must ensure that there is no pregnancy. Patients should be informed that as soon as a missed period occurs or if there is any other reason to suspect pregnancy, they should immediately inform the doctor about this in order to take a pregnancy test. In the case of a positive pregnancy test, the doctor should discuss with the patient the possible risks to which the pregnancy is exposed. It is possible that rapid reduction of the active metabolite in the blood using the drug elimination procedure described below will help reduce the risk to the fetus from leflunomide during the first missed period. Women who are taking leflunomide and wish to become pregnant are advised to follow one of the following procedures to ensure that the fetus is not exposed to toxic concentrations of A771726 (reference concentration below 0.02 mg/L). Waiting period. It can be expected that the plasma concentration of A771726 may be higher than 0.02 mg/L for a long period. It is believed that its concentration may become less than 0.02 mg/l 2 years after stopping treatment with leflunomide. The first time the concentration of A771726 in blood plasma is measured after a two-year waiting period. After that, it is necessary to measure the concentration of A771726 in blood plasma, at least after 14 days. If the value of both measurements is below 0.02 mg/l, no teratogenic risk is expected. The procedure of "laundering". Colorumin 8 g is introduced 3 times a day for 11 days; As an alternative to 50 g of activated coal chopped into powder, is introduced 4 times a day for 11 days. Regardless of the selected “laundering” procedure, it is necessary to check with two separate tests with the interval of at least 14 days and wait a month and a half from the moment when the concentration of the drug in the plasma will be recorded for the first time below 0.02 mg/l, until fertilization. It is necessary to inform women of the childbearing period that 2 years should pass after the cessation of treatment with leflunomide before they may try to get pregnant. If the 2-year waiting period with reliable contraception seems unreasonable, it is possible to advise the procedure for “laundering” for preventive purposes. Both colestyramine and activated carbon can affect the absorption of estrogens and progestogen, therefore reliable oral contraceptives do not give one hundred percent guarantees during the period of “washing” with a coal or activated coal. It is recommended to use alternative contraceptive methods. Animal studies have shown that leflunomide or its metabolites pass into breast milk, so women who breastfeed should not take the drug. Leflunomide can be used in patients only after a thorough medical examination. The procedure of "laundering". The “laundering” procedure is carried out according to the following scheme: -Kolestiramin 8 g is introduced 3 times a day for 11 days; -in the quality of alternatives -50 g of activated coal chopped into powder, is introduced 4 times a day for 11 days. Impact on the ability to drive vehicles and machinery. Taking the drug may be accompanied by headache, dizziness. In this regard, patients taking leflunomide should be cautioned when managing dangerous mechanical agents, including a car. Storage conditions Store in a dry place, protected from light, out of reach of children at a temperature not exceeding 25 C.
Drug interactions Leflunomide
There is currently no information regarding the combined use of leflunomide with antimalarial drugs used in rheumatology (for example, chloroquine and hydroxychloroquine), gold preparations (IM or orally), D-penicillamine, azathioprine and other immunosuppressive drugs (except methotrexate). The risk associated with the prescription of complex therapy, especially with long-term treatment, is unknown. Since this type of therapy can lead to the development of additive or even synergistic toxicity (for example, hepato- or hematotoxicity), combinations of this drug with other basic drugs (for example, methotrexate) are undesirable. Recent, concomitant, or subsequent use of potentially myelotoxic agents may be associated with a greater risk of hematologic effects. Immunosuppressants increase the risk of developing infections, as well as malignancies, especially lymphoproliferative diseases. Caution should be exercised when prescribing drugs metabolized by CYP 2C9 (phenytoin, warfarin, tolbutamide), with the exception of NSAIDs. Increased severity of adverse events may occur when recent or concomitant use of hepatotoxic or hematotoxic drugs is used or when these drugs are started after treatment with leflunomide without a washout period. In patients with rheumatoid arthritis, no pharmacokinetic interaction was found between leflunomide (10–20 mg/day) and methotrexate (10–25 mg/week). Cholestyramine or activated carbon quickly and significantly reduce the concentration of A771726 in the blood plasma. Possible combined use with NSAIDs and GCS. The enzymes involved in the metabolism of leflunomide and its metabolites are not precisely known. There is no clinically significant interaction with cimetidine (a nonspecific inhibitor of cytochrome P450). Following concomitant administration of a single dose of leflunomide with multiple doses of rifampicin (a nonspecific cytochrome P450 inducer), maximum concentrations of A771726 increased by approximately 40%, whereas AUC did not change significantly. The mechanism of this effect is not clear. There is no decrease in the contraceptive effect when used together with triphasic oral contraceptives containing 30 mcg ethinyl estradiol, while the pharmacokinetics of A771726 did not change. There is no data regarding the effectiveness and safety of vaccination during treatment with leflunomide (vaccination with live vaccines is not recommended). The long half-life of the drug should be taken into account when planning vaccination with a live vaccine after its discontinuation.
Teriflunomide
special instructions
Treatment should be carried out under the supervision of a physician experienced in treating patients with multiple sclerosis. Before starting treatment, the following studies should be carried out: measuring blood pressure, determining ALT activity, a complete blood count, including a leukocyte count and determining the number of platelets in the blood.
During treatment with teriflunomide, the following parameters should be regularly monitored: blood pressure, ALT activity.
If new symptoms and signs (for example, infection) appear during treatment, a complete blood count, including a leukocyte count, and a determination of the platelet count in the blood should be performed
Teriflunomide is slowly cleared from plasma: plasma concentrations reach values below 0.02 mg/l on average within 8 months, although due to individual deviations in the process of drug elimination it can last up to 2 years. The elimination of the drug can be accelerated through an accelerated elimination procedure. The accelerated elimination procedure may be used at any time after stopping teriflunomide.
An increase in liver enzyme activity was observed in patients taking teriflunomide. These adverse reactions occurred mainly in the first 6 months of treatment.
Patients with a history of liver disease are at risk of worsening liver function while taking teriflunomide. In this group of patients, symptoms of liver damage should be carefully monitored.
Teriflunomide should be prescribed with caution to patients who abuse alcohol.
Because teriflunomide is highly bound to plasma proteins, mainly albumin, plasma concentrations of unbound teriflunomide may be increased in patients with hypoproteinemia, such as those with nephrotic syndrome. Teriflunomide should not be prescribed to patients with severe hypoproteinemia.
If blood pressure increases, appropriate antihypertensive therapy should be administered before and during treatment with teriflunomide.
Initiation of treatment with teriflunomide should be delayed in patients with serious active infections until complete recovery.
Given the immunomodulatory effect of teriflunomide, if a patient develops a serious infection, the need to pause treatment with the drug should be considered and the possible benefits and risks assessed before restarting detoxification therapy. Due to the long T1/2, it is necessary to consider the need for accelerated elimination using cholestyramine or activated carbon.
Patients who test positive for tuberculosis at screening should receive appropriate treatment before starting teriflunomide.
During therapy, interstitial lung diseases can develop acutely.
Pulmonary symptoms, such as persistent cough and shortness of breath, may prompt discontinuation of therapy and further evaluation. In patients with existing anemia, leukopenia and/or thrombocytopenia, as well as in patients with impaired bone marrow function or at risk of suppression of bone marrow hematopoiesis, the risk of hematological diseases during teriflunomide therapy is increased. In the event of the development of these adverse reactions, it is necessary to consider the possibility of using an accelerated elimination procedure to reduce the concentration of teriflunomide in plasma. In cases of severe hematological reactions, including pancytopenia, teriflunomide and any other myelosuppressive drug should be discontinued. The feasibility of an accelerated elimination procedure should be considered.
If ulcerative stomatitis occurs, teriflunomide should be discontinued. If severe generalized skin reactions (Steven-Johnson syndrome or toxic epidermal necrolysis - Lyell's syndrome) are suspected when skin and/or mucocutaneous reactions occur, teriflunomide and any other drugs potentially causing such reactions should be stopped and should be started immediately. accelerated elimination procedure. In such cases, patients should not be reintroduced to teriflunomide. If peripheral neuropathy is diagnosed in a patient taking teriflunomide, discontinuation of teriflunomide and an accelerated elimination procedure should be considered. The use of live attenuated vaccines may pose a risk of infection and should therefore be avoided. Because leflunomide is the parent compound of teriflunomide, coadministration of teriflunomide with leflunomide is not recommended.
Switching to or from teriflunomide Based on clinical data relating to concomitant use of teriflunomide with interferon beta or glatiramer acetate, there is no need for a waiting period when initiating teriflunomide therapy after interferon beta or glatiramer acetate, or when initiating interferon beta therapy. or glatiramer acetate after teriflunomide.
Due to the long T1/2 of natalizumab, concurrent exposure and, therefore, concomitant effects on the immune system may occur if teriflunomide therapy is initiated within 2-3 months after discontinuation of natalizumab. Therefore, precautions should be taken when switching from natalizumab therapy to teriflunomide.
Taking into account the T1/2 of fingolimod, a 6-week interval without therapy is required to eliminate circulating substances from the body. It takes 1 to 2 months for the lymphocyte count to return to normal after stopping fingolimod. This may result in additive effects on the immune system. Therefore, precautions should be taken when switching from fingolimod therapy to teriflunomide.
In multiple sclerosis, the median T1/2 was approximately 19 days after repeated doses of 14 mg. If the decision is made to stop treatment with teriflunomide within 5 T1/2 (approximately 3.5 months, although may be longer in some patients), starting other therapy will result in concurrent exposure to teriflunomide. This may lead to additive effects on the immune system, which requires careful precautions.
Impact on the ability to drive vehicles and machinery
If undesirable effects from the nervous system occur, for example, dizziness, you should refrain from driving vehicles and engaging in other potentially hazardous activities.
Leflunomide overdose, symptoms and treatment
There are no data regarding overdose of leflunomide or intoxication caused by it in humans. The use of cholestyramine or activated carbon is indicated. Cholestyramine, taken orally 8 g 3 times a day for 24 hours, reduces the content of A771726 in the blood plasma by approximately 40% after 24 hours and by 49–65% after 48 hours. Administration of activated carbon (in the form of a suspension) orally or through the gastric the probe (50 g every 6 hours during the day) reduces the concentration of the active metabolite A771726 in plasma by 37% after 24 hours and by 48% after 48 hours.
List of pharmacies where you can buy Leflunomide:
- Moscow
- Saint Petersburg