Фенилкетонурия
(ФКУ) ― наследственное нарушение метаболизма аминокислот, в первую очередь фенилаланина (ФА), входящего в состав белков. Вещество участвует в укладке белка и стабилизации белковых структур.
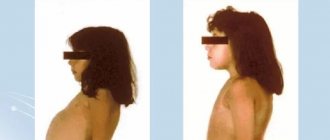
Первые симптомы: частое срыгивание, рвота, экземы, судороги, исходящий от мочи и кожи запах плесени. Ребенок может был вялым либо, наоборот, гиперактивным. Отстает в психомоторном развитии, наблюдаются признаки олигофрении. Диагноз может быть поставлен в родильном доме. Все дети с фенилкетонурией безусловно получают статус «ребенок-инвалид».
Лечение заболевания заключено в соблюдении специальной низкобелковой диете, не содержащей продукты с ФА.
Определение заболевания
Фенилкетонурия ― это врожденная, генетическая патология, подразумевающая нарушения гидроксилирования фенилаланина. Характеризуется накоплением в организме аминокислоты и продуктов ее метаболизма, что ведет к тяжелым поражениям центральной нервной системы. Впервые заболевание было описано норвежским врачом И. А. Феллингом в 1934 году.
Изучая болезнь специалисты установили, что за наличие болезни отвечает единственный ген фенилаланингидроксилазы. Первое успешное лечение было разработано и проведено в 1950 году в Англии.
В неонатальном периоде клиника отсутствует. Патология проявляется в первые полгода жизни ребенка. В дальнейшем накопление вещества приводит к тяжелым нарушениям развития. Поэтому крайне важно сразу после рождения выявить дефект и не допустить употребление продуктов, содержащих фенилаланин. Более позднее соблюдение диеты не устранит полученные нарушения, но не допустит развития новых.
Патология одинаково часто встречается среди лиц обоих полов. Расовых особенностей не выявлено. Большое количество больных в таких странах как Китай, Турция, Ирландия. В среднем по России с фенилкетонурией рождается каждый 7-ми тысячный ребенок.
История
Открыл фенилкетонурию в 1934 году норвежский доктор Ивар Асбьерн Феллинг. Положительный исход лечения впервые наблюдался в Великобритании (в Бирмингемском госпитале для детей) благодаря стараниям коллектива врачей во главе с Хорстом Биккелем в первой половине 50-х годов XX века. Однако действительно большой успех в лечении этой болезни был отмечен в 1958-1961 годы, когда появились первые методы анализа крови младенцев на содержание в ней высоких концентраций фенилаланина, говорящих о наличии недуга.
Выяснилось, что за развитие болезни отвечает всего лишь один ген, получивший наименование РАН (ген фенилаланингидроксилазы).
Благодаря этой находке ученым и докторам по всему миру удалось более подробно выделить и описать как само заболевание, так и его симптомы и формы. Более того, были найдены и разработаны абсолютно новые, высокотехнологичные и современные способы лечения, такие, как генотерапия, которая на сегодня является образцом эффективной борьбы с генетическими патологиями человека.
Причины фенилкетонурии
Существует три типа генетического отклонения, первый считается классическим, поскольку диагностируется в более чем 90% случаев. Второй и третий ― более редкая форма патологии. Симптоматика схожа во всех типах, заболевание приводит к умственной отсталости. При классической форме фенилкетонурии избежать этого можно диетотерапией, но атипичные варианты, к сожалению, коррекции не подлежат.
- Классическая фенилкетонурия (I тип) ― это низкая выработка фенилаланингидроксилазы (ФАГ), что приводит к собиранию в естественных жидкостях организма фенилаланина и продуктов его расщепления. Патология вызвана мутированным геном РАН.
- Фенилкетонурия II типа ― недостаток дигидроптеридинредуктазы, что препятствует преобразованию фенилаланина в тирозин. Патология из-за мутации гена QDPR.
- Фенилкетонурия III типа ― недостаток 6-пирувоилтетрагидроптеринсинтазы, нужной для синтеза тетрагидробиоптерина. Патология вызвана мутированным PTS-геном.
Все формы заболевания наследуются по аутосомно-рецессивной форме. Это означает, что генетический дефект может быть унаследован у одного из родителей. Половая принадлежность родителя и ребенка не играет роли.
Фенилкетонурия у детей и ее лечение
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотипов ФКУ [1].
Определение, патогенез, классификация
Фенилкетонурия — наследственная аминоацидопатия, связанная с нарушением метаболизма фенилаланина, в результате мутационной блокады ферментов приводящая к стойкой хронической интоксикации и поражению ЦНС c выраженным снижением интеллекта и неврологическим дефицитом [1, 2].
Основное значение в патогенезе классической ФКУ имеет неспособность фенилаланингидроксилазы перерабатывать фенилаланин до тирозина. В результате в организме накапливается фенилаланин и продукты его аномального обмена (фенилпировиноградная, фенилуксусная, фенилмолочная кислоты) [1–3].
В числе других патогенетических факторов рассматриваются нарушения аминокислотного транспорта через гематоэнцефалический барьер, нарушения церебрального пула аминокислот с последующим нарушением синтеза протеолипидных белков, нарушения миелинизации, низкие уровни нейротрансмиттеров (серотонин и др.) [1–4].
Фенилкетонурия I (классическая или тяжелая) — аутосомно-рецессивное заболевание, вызванное мутацией гена фенилаланингидроксилазы (длинное плечо хромосомы 12); выявлены 12 различных гаплотипов, из которых около 90% ФКУ ассоциировано с четырьмя гаплотипами. Наиболее частые мутации в гене фенилаланингидроксилазы: R408W, R261Q, IVS10 nt 546, Y414C. В основе болезни — дефицит фенилаланин-4-гидроксилазы, обеспечивающей конверсию фенилаланина в тирозин, что приводит к накоплению в тканях и физиологических жидкостях фенилаланина и его метаболитов [1–4].
Особую группу составляют атипичные варианты ФКУ, при которых клиническая картина напоминает классическую форму болезни, но по показателям развития, несмотря на проведение диетотерапии, не отмечается положительной динамики. Эти варианты ФКУ связаны с дефицитом тетрагидроптерина, дегидроптеринредуктазы, 6-пирувоилтетрагидроптеринсинтазы, гуанозин-5-трифосфатциклогидролазы и т. д. [1–4].
Фенилкетонурия II (атипичная) — аутосомно-рецессивное заболевание, при котором генный дефект локализуется в коротком плече хромосомы 4 (участок 4р15.3), характеризующееся недостаточностью дегидроптеринредуктазы, приводящей к нарушению восстановления активной формы тетрагидробиоптерина (кофактор в гидроксилировании фенилаланина, тирозина и триптофана) в сочетании со снижением в сыворотке крови и спинномозговой жидкости фолатов. Результатом являются метаболические блоки в механизмах превращения фенилаланина в тирозин, а также предшественников нейромедиаторов катехоламинового и серотонинового рядов (L-дофа, 5-окситриптофан). Болезнь описана в 1974 г. [1–4].
Фенилкетонурия III (атипичная) — аутосомно-рецессивное заболевание, связанное с недостаточностью 6-пирувоилтетрагидроптеринсинтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптеринтрифосфата (описано в 1978 г.). Дефицит тетрагидробиоптерина приводит к расстройствам, сходным с нарушениями при ФКУ II [1–4].
Примаптеринурия — атипичная ФКУ у детей с легкой гиперфенилаланинемией, у которых в моче в больших количествах присутствует примаптерин и некоторые его производные при наличии нормальной концентрации в спинномозговой жидкости нейромедиаторных метаболитов (гомованилиновой и 5-оксииндолуксусной кислот). Энзиматический дефект пока не выявлен [1–4].
Материнская ФКУ — заболевание, сопровождающееся снижением уровня интеллекта (до умственной отсталости) среди потомства женщин, страдающих ФКУ и не получающих специализированную диету в совершеннолетнем возрасте. Патогенез материнской ФКУ детально не изучен, но предполагается ведущая роль хронической интоксикации плода фенилаланином и продуктами его аномального метаболизма [1–4].
R. Koch и соавт. (2008) при аутопсии головного мозга младенца, у матери которого отмечалась ФКУ (без адекватного контроля за уровнем фенилаланина в крови), обнаружили ряд патологических изменений: низкий вес мозга, вентикуломегалию, гипоплазию белого вещества и задержку миелинизации (без признаков астроцитоза); хронических изменений в сером веществе головного мозга не было обнаружено. Предполагается, что нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита при материнской ФКУ [5].
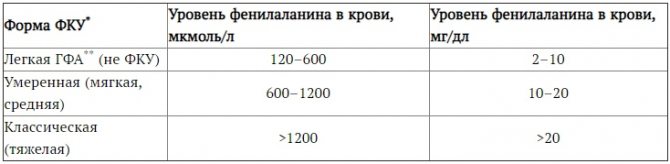
В практических целях в медико-генетических центрах РФ используется условная классификация ФКУ, основанная на уровнях содержания фенилаланина в сыворотке крови: классическая (тяжелая или типичная) — уровень фенилаланина выше 20 мг% (1200 мкмоль/л); средняя — 10,1–20 мг% (600–1200 мкмоль/л), а также уровень фенилаланина 8,1–10 мг%, если он устойчив на фоне физиологической нормы потребления белка в рационе питания; легкая (гиперфенилаланинемия, не требующая лечения) — уровень фенилаланина до 8 мг% (480 мкмоль/л) [2].
Клинические проявления и диагностика
При рождении дети с ФКУ I выглядят здоровыми, хотя чаще имеется специфический хабитус (светлые волосы, голубые глаза, суховатая кожа). При отсутствии своевременного выявления и лечения болезни в течение первых двух месяцев жизни у них появляется частая и интенсивная рвота и повышенная раздражительность. Между 4 и 9 месяцами становится очевидным выраженное отставание в психомоторном развитии [1–4].
Пациентов отличает специфический («мышиный») запах кожных покровов. Выраженные неврологические нарушения у них редки, но характерны черты гиперактивности и расстройств аутистического спектра. При отсутствии своевременного лечения уровень IQ составляет < 50. Судорожные приступы, характерные для детей с выраженным интеллектуальным дефицитом, чаще дебютируют в возрасте до 18 месяцев (могут исчезать спонтанно). В раннем возрасте приступы нередко имеют форму инфантильных спазмов, впоследствии трансформируясь в тоникоклонические припадки [1–4].
Из диагностических методов (помимо определения содержания в крови уровней фенилаланина и тирозина), используются проба Феллинга, тест Гатри, хроматография, флуориметрия, поиск мутантного гена. Широко применяются ЭЭГ и МРТ-исследование [1–5].
При ЭЭГ выявляются нарушения, преимущественно в виде паттерна гипсартимии (даже при отсутствии приступов); типичны единичные и множественные фокусы спайк- и полиспайк-разрядов [1–4].
МРТ-данные обычно аномальны вне зависимости от проведения/отсутствия лечения ФКУ: на Т2-взвешенном изображении имеется повышение интенсивности сигнала в перивентрикулярном и субкортикальном белом веществе задних отделов гемисфер. Хотя у детей может отмечаться кортикальная атрофия, изменений сигнала в стволе, мозжечке или коре головного мозга не обнаруживается. Описанные изменения при МРТ-исследовании не соотносятся с уровнем IQ, но зависят от уровня содержания фенилаланина в крови [1–4].
При фенилкетонурии II у пациентов клиническая симптоматика появляется в начале второго года жизни. Несмотря на проведение диетотерапии, назначаемой после выявления повышенного уровня содержания в крови фенилаланина в периоде новорожденности, отмечается прогрессирующее течение болезни. Имеется выраженная умственная отсталость, признаки повышенной возбудимости, судороги, мышечная дистония, гиперрефлексия (сухожильная) и спастический тетрапарез. Нередко к 2–3-летнему возрасту наступает летальный исход [1–4].
Клиническая картина фенилкетонурии III напоминает таковую ФКУ II; она включает следующую триаду признаков: глубокая умственная отсталость, микроцефалия, спастический тетрапарез [1–4].
Профилактика
Необходимо своевременное выявление ФКУ с использованием соответствующих скрининг-тестов в родильных домах, а также генетическое консультирование. Будущим матерям c ФКУ для предотвращения повреждения плода до зачатия и на протяжении всей беременности рекомендуется строго соблюдать диету с низким содержанием фенилаланина, поддерживая его уровень < 4 мг% (< 242 мкмоль/л). Потомство матерей с легкой ФКУ (фенилаланин < 6,6 мг% или < 400 мкмоль/л) не страдает [1–4].
Новые методы лечения
В настоящее время интенсивно разрабатываются сразу несколько видов альтернативной терапии ФКУ. Среди них: так называемый метод «больших нейтральных аминокислот» (large neutral amino acids), энзимотерапия фенилаланингидроксилазой, фенилаланинаммониалиазой; лечение тетрагидробиоптерином (Сапроптерин) [6–11].
Есть данные об успешном лечении пациентов с умеренной или легкой ФКУ с применением тетрагидробиоптерина (10–20 мг/кг/сут) [7, 9, 10].
D. M. Ney и соавт. (2008) показали, что использование пищевых гликомакропептидов при ФКУ (с ограниченной дотацией незаменимых кислот) снижает концентрации фенилаланина в плазме крови и головном мозге, а также способствует адекватному физическому развитию [8]. Экспериментальным методом лечения ФКУ является введение гена фенилаланингидроксилазы непосредственно в пораженные клетки печени. В РФ указанные методы в настоящее время не применяются.
Диетотерапия
Именно лечебная диета максимально эффективна в предотвращении интеллектуального дефицита при тяжелой (классической) ФКУ. Наибольшее значение имеет возраст пациента к моменту начала диетотерапии (IQ снижается примерно на 4 балла за каждый месяц от рождения до начала лечения). Подходы к диетотерапии ФКУ в разных странах несколько различаются, но сами их принципы являются согласованными [1–4].
Диетические ограничения не показаны младенцам, у которых уровень фенилаланина в крови находится в пределах 2–6 мг% (120–360 мкмоль/л). Основа диеты при ФКУ — назначение рационов питания с низким содержанием фенилаланина, источником которого является белковая пища. Такая диета назначается всем пациентам первого года жизни. Она должна быть назначена детям с выявленной ФКУ до 8-недельного возраста; ее применение в более позднем возрасте гораздо менее эффективно [1–4].
Общая характеристика диеты при ФКУ. Лечебный рацион питания при ФКУ представлен тремя основными компонентами: лечебные продукты (смеси аминокислот без фенилаланина), натуральные продукты питания (подобранные), малобелковые продукты на основе крахмала.
Из рациона питания при ФКУ исключают продукты животного происхождения с высоким содержанием белка (мясо, птица, рыба, молочные продукты и т. д.). Грудное молоко на первом году жизни ограничивается (ранее отменялось полностью). Из смесей (заменителей грудного молока) предпочтение отдается содержащим меньшее количество белка [1–4].
Диетотерапия на первом году жизни. Эквивалентную замену по белку и фенилаланину производят с использованием «порционного» способа расчета: 50 мг фенилаланина приравниваются к 1 г белка (для адекватной замены продуктов по белку и фенилаланину). Так как фенилаланин является эссенциальной аминокислотой, для обеспечения нормального развития ребенка с ФКУ должна быть удовлетворена минимальная потребность в ней. В течение первого года жизни допустимое количество фенилаланина составляет от 90 до 35 мг/кг ребенка [1–4].
Для детей с ФКУ в возрасте до 12 месяцев в РФ в настоящее время представлены следующие лечебные продукты зарубежного и отечественного производства: Афенилак (РФ), MD мил ФКУ-0 (Испания) и ХР Аналог LCP (Нидерланды-Великобритания).
Диетотерапию начинают при уровне фенилаланина в крови от 360–480 ммоль/л и выше. Именно показатель его содержания в крови считается главным критерием диагностики и оценки эффективности проводимого лечения.
Введение прикорма и дополнительных продуктов питания. После трех месяцев рацион питания начинают расширять за счет использования соков (фруктовых и ягодных), назначая их с 3–5 капель, с постепенным увеличением объема до 30–50 мл, а к концу первого года жизни — до 100 мл. Основные соки: яблочный, грушевый, сливовый и т. д. Фруктовые пюре назначают, увеличивая их количество в рационе питания аналогично с таковым вводимого сока [2].
В период с 4–4,5 месяцев в рацион питания вводят первый прикорм в виде овощного пюре, приготовленного самостоятельно (или плодоовощных консервов для питания детей грудного возраста — последние без добавления молока). Далее последовательно назначается 2-й прикорм — каша (10%) из молотого саго или безбелковой крупки. Могут быть использованы безмолочные каши промышленного производства на основе кукурузной и/или рисовой муки, содержащие не более 1 г белка в 100 мл готового к употреблению продукта.
После 6 месяцев в питание можно ввести кисели и/или муссы (безбелковые), которые готовятся с использованием амилопектинового набухающего крахмала и фруктового сока, безбелковый напиток с молочным вкусом Нутриген или низкобелковый молочный напиток PKU «Лопрофин».
С 7 месяцев ребенок с ФКУ может получать низкобелковые изделия «Лопрофин», например, спиральки, спагетти, рис или безбелковую вермишель, а с 8 месяцев — специальный безбелковый хлеб [2].
Диетотерапия у детей старше одного года. Особенности составления лечебных диет для пациентов старше 12 месяцев заключаются в использовании продуктов на основе смесей аминокислот без фенилаланина и/или гидролизатов белка с незначительным его количеством (превышающим таковое в продуктах для детей с ФКУ первого года жизни), в состав которых введены комплексы витаминов, макро- и микроэлементов. Доля белкового эквивалента по мере роста детей постепенно увеличивается, а квота жирового и углеводного компонентов, наоборот, снижается (в дальнейшем — полностью исключается), что позволяет впоследствии значительно расширить рацион пациентов за счет подобранных натуральных продуктов [1–4].
Количество фенилаланина, которое детям различного возраста разрешается получать алиментарным путем при следовании лечебной диете, постепенно снижается с 35 до 10 мг/кг/сут [1–4].
В диетотерапии детей старше одного года принято использовать специализированные лечебные продукты (на основе смесей аминокислот без фенилаланина): Тетрафен 30, Тетрафен 40, Тетрафен 70, MD мил ФКУ-1, MD мил ФКУ-3 (Испания).
Особым разнообразием и проверенным годами качеством отличаются продукты «Нутриции» (Нидерланды–Великобритания): П-АМ 1, П-АМ 2, П-АМ 3, Изифен (готовый к употреблению продукт), а также ХР Максамейд и ХР Максамум с нейтральным и апельсиновыми вкусами.
Рекомендуется осуществлять постепенный переход со специализированной смеси (для грудного возраста) на продукты для детей более старшего возраста постепенно (в течение 1–2 недель). При этом объем предыдущей смеси уменьшают на 1/4–1/5 часть и добавляют эквивалентное по белку количество нового продукта. Новый лечебный продукт (количество которого рассчитывают в зависимости от массы тела и допустимых по возрасту количеств фенилаланина) предпочтительно давать ребенку дробно, 3–4 раза в день, предлагая его запивать соками, водой или другими напитками [2].
Ассортимент продуктов для детей с ФКУ существенно ограничен. В периоде максимально строгого соблюдения диеты (грудной и ранний детский возраст) обязательно использование специализированных лечебных продуктов. Целью их применения при ФКУ является замещение источников белка при полном соответствии нормам потребления основных нутриентов детьми (с учетом возраста и конкретной клинической ситуации). Часть лечебных продуктов содержит полиненасыщенные жирные кислоты (омега-6 и омега-3) в соотношении 5:1–10:1; таким источникам пищи отдается предпочтение [1–4].
Из специальных продуктов применяются сухие аминокислотные смеси, лишенные содержания фенилаланина, с дотацией белкового эквивалента — его артифициального аналога (в количествах, соответствующих возрасту больных ФКУ).
Другие доступные в РФ малобелковые продукты для диетотерапии ФКУ включают саго, специальный хлеб, вермишель и другие виды лечебного питания [2, 3]. Эти лечебные продукты (амилофены) основаны на крахмалах, не содержащих трудноусвояемых углеводов и минеральных веществ. Они представлены макаронными изделиями, крупами, саго, специальной мукой, хлебобулочными изделиями, инстантами для приготовления киселей, муссов и т. д. Витаминные добавки повышают питательную ценность продуктов с низким содержанием белков [2].
Существуют также малобелковые продукты зарубежного производства, Лопрофины (Нидерланды-Великобритания), на основе крахмалов (пшеничного, рисового, картофельного, кукурузного и т. д.), включающие макаронные изделия, крупы для приготовления каш, специальные сорта хлеба (из тапиоки, пшеничного и рисового крахмала), печенье, крекеры, сухари, а также мука, различные дессерты, приправы и соусы с привлекательным вкусом, значительный ассортимент напитков (включая заменители молока, сливок и кофе) и т. д. [3].
Расчет и составление диеты. Используется следующая формула: А = В + С, где А — общая потребность в белках, В — белок натуральной пищи, С — белок, обеспечиваемый за счет лечебных продуктов питания [1–4].
Обогащение диеты тирозином. Некоторые исследователи предлагают обогащать диету с низким содержанием фенилаланина тирозином, хотя при этом отсутствует статистически достоверное подтверждение лучшего интеллектуального развития при следовании ФКУ-рациону [11].
Органолептические свойства диеты. Вкусовые свойства практически всех искусственных лечебных продуктов для пациентов с ФКУ специфические. Для маскировки органолептически неприятных качеств лечебной диеты при ФКУ используют различные вкусовые добавки (лишенные белка) и специальные рецептуры. Подсластитель аспартам не должен использоваться, так как расщепляется до фенилаланина, метанола и аспартата [2].
Контроль эффективности диетотерапии. Основан на регулярном контроле содержания фенилаланина в крови (он должен находиться в средних пределах 3–4 мг% или 180–240 мкмоль/л) [1–4].
В РФ используется следующая схема контроля за содержанием фенилаланина в крови у пациентов с ФКУ: до 3-месячного возраста — 1 раз в неделю (до получения стабильных результатов) и далее не менее 2 раз в месяц; от 3 месяцев до 1 года — 1 раз в месяц (при необходимости — 2 раза в месяц); от 1 года до 3 лет — не менее 1 раза в 2 месяца; после 3 лет — 1 раз в 3 месяца [2].
Постоянно контролируется нутритивный статус больного, его физическое и интеллектуальное, эмоциональное и речевое развитие. При необходимости к обследованию больного привлекаются врачи-специалисты, проводится психолого-дефектологическое тестирование и ряд исследований (УЗИ внутренних органов, ЭКГ, ЭЭГ, МРТ головного мозга, общий анализ крови и мочи, протеинограмма крови, по показаниям — глюкоза, холестерин, креатинин, ферритин, сывороточное железо и др.). Общий анализ крови проводится с частотой 1 раз в месяц, биохимическое исследование крови — по показаниям [1–4].
Питание при инфекционных заболеваниях. При интеркуррентных заболеваниях с гипертермией, интоксикацией и/или диспепсическими явлениями возможно временное прекращение диетотерапии (на несколько дней) с заменой лечебных продуктов на натуральные (с невысоким содержанием белка). По завершении острого периода заболевания лечебный продукт снова вводится в рацион питания, но за менее продолжительный период, чем в начале диетотерапии [2].
Прекращение диетотерапии. Дискутабельным моментом продолжает оставаться возраст пациентов с ФКУ, в котором можно прекращать диетотерапию.
Есть данные, что при отмене диетотерапии в 5-летнем возрасте у одной трети детей с ФКУ отмечалось снижение уровня IQ на 10 баллов и более в течение последующих 5 лет. У пациентов старше 15 лет перерывы в диетотерапии нередко сопровождаются прогрессирующими изменениями белого вещества мозга (по данным МРТ) [1].
Диетотерапия пациентов с классической ФКУ должна быть пожизненной [1–4].
В РФ, в соответствии с законодательством, специальная диетотерапия должна предоставляться пациенту бесплатно, независимо от степени инвалидизации и возраста пациента. Строгое, обязательное диетическое лечение ФКУ проводится обычно до 18-летнего возраста с последующим расширением рациона. Совершеннолетним пациентам рекомендуется отказаться от потребления высокобелковых продуктов животного происхождения (общее количество белка при этом не должно превышать 0,8–1,0 г/кг/сут).
Литература
- Blau N. et al. Phenylketonuria // Lancet. 2010. № 376. P. 1417–1427.
- Лечебное питание при наследственных нарушениях обмена (Е70.0-Е74.2). В кн.: Клиническая диетология детского возраста / Под ред. Боровик Т. Э., Ладодо К. С. М.: «МИА». 2008. С. 330–383.
- Harding C. O. et al. Advances and challenges in phenylketonuria // J. Inherit. Metab. Dis. 2010. V. 33. P. 645–648.
- Lord B. et al. Implications of resolving the diagnosis of PKU for parents and children // J. Pediatr. Psychol. 2008. V. 33. P. 855–866.
- Koch R. et al. Neuropathology of a 4-month-old infant born to a woman with phenylketonuria // Dev. Med. Child. Neurol. 2008. V. 50. P. 230–233.
- Van Spronsen F. J. et al. Large neutral amino acids in the treatment of PKU: from theory to practice // J. Inherit. Metab. Dis. 2010. V. 33. P. 671–676.
- Harding C. O. New era in treatment for phenylketonuria: pharmacologic therapy with sapropterin dihydrochloride // Biologics. 2010. V. 9. P. 231–236.
- Ney D. M. et al. Dietary glycomacropeptide supports growth and reduces the concentrations of phenylalanine in plasma and brain in a murine model of phenylketonuria // J. Nutr. 2008. V. 138. P. 316–322.
- Singh R. H. et al. BH4 therapy impacts the nutrition status and intake in children with phenylketonuria: 2-year follow-up // J. Inherit. Metab. Dis. 2010. V. 33. P. 689–695.
- Trefz F. K. et al. Sapropterin dihydrochloride: a new drug and a new concept in the management of phenylketonuria // Drugs Today. 2010. V. 46. P. 589–600.
- Webster D. et al. Tyrosine supplementation for phenylketonuria // Cochrane Database Syst. Rev. 2010. V. 8: CD001507.
В. М. Студеникин, доктор медицинских наук, профессор Т. Э. Боровик, доктор медицинских наук, профессор Т. В. Бушуева, кандидат медицинских наук
НЦЗД РАМН, Москва
Контактная информация об авторах для переписки
Классификация
Фенилкетонурия в настоящее время не имеет общепризнанной мировой классификации. Над этим вопросом ведутся дебаты, наравне с изучением заболевания. Чуть ранее, до расшифровки генов, считалось, что степень поражения интеллектуальных способностей зависит от степени активности фермента. Поэтому текущая квалификация признана устаревшей. Не учитывает она и другие симптоматические факторы.
При диагностировании ставят:
- I тип (дефицит ФАГ) ― концентрация ФА больше 20 мг/дл.
- Средняя форма ФКУ ― ФА от 8,1 до 20 мг/дл.
- Легкая форма ГФА-уровень ― ФА от 2,1 до 8,0 мг/дл.
При уровне до 8,0 мг/дл фенилкетонурию считают доброкачественной. Она не требует специального лечения, но необходимо наблюдение первый год жизни ребенка. Контролирует состояние врач-педиатр, невролог, генетик.
Выделяют также еще одну форму фенилкетонурии, не требующую коррекции. Это транзиторная форма ГФА в период новорожденности. Возникает, как правило, при недоношенности, что обусловлено функциональной незрелостью организма. Транзиторная фенилкетонурия ― это временное повышение ФА-уровня, способное подняться до критических значений. При этом клиника отсутствует либо проявления весьма незначительны. Через несколько месяцев биохимические показатели приходя в норму.
Патогенез
Механизм зарождения и развития фенилкетонурии связан с нарушением обмена органического соединения ― аминокислоты фенилаланина. Метаболический блок препятствует преобразованию фенилаланина в тирозин. Аминокислота не только не преобразуется, а накапливается в виде токсичных метаболитов:
- фенилмолочная кислота;
- фенилпировиноградная кислота;
- фенилуксусная кислота;
- фенилэтиламин и прочее.
Скопление фенил-веществ оказывает токсическое действие на ЦНС. В настоящий момент механизм еще до конца не изучен, врачам не известен патогенез дисфункции головного мозга.
Существуют предположения, что поражение нервной системы является результатом ряда факторов. Среди них как прямое токсического воздействие фенилаланина, так и нарушение обмена белков, липопротеидов и гликопротеидов, сбой гомонального метаболизмеа и мембранного транспорта аминокислот. Все это в комплексе имеет важное значение для созревания и правильного функционирования ЦНС.
Симптомы
I тип. Первые признаки у ребенка проявляются в возрасте от 2 месяцев до полугода.
- Апатичность либо, наоборот, повышенная раздражительность.
- Отсутствие интереса к окружению, людям, предметам, обстановке.
- Частое срыгивание.
- Аллергический дерматит.
- Нарушение мышечного тонуса.
- Пониженное давление.
- Судороги.
- Иногда развивается микроцефалия (малый размер черепа относительно других частей тела) и гидроцефалия (избыточная жидкость, омывающая головной мозг).
К характерным симптомам относятся гипопигментация кожи, волос, радужной оболочки глаз. Моча имеет специфический запах плесени или его еще называют «мышиным» запахом. Эпилептические припадки наблюдаются у половины больных, часто является первым выраженным клиническим симптомом. Приступ характеризуется «салаамовыми» судорогами, напоминающими кивки. Они случаются часто, плохо поддаются антиконвульсантному лечению.
Если не корректировать концентрацию ФА, болезнь прогрессирует. Как правило, уровень IQ у таких детей не превышает 20, при норме от 85. Умственная отсталость настолько сильная, что отсутствуют эмоциональные реакции, наблюдаются психопатии и шизофреноподобные расстройства.
II тип. Первая симптоматика проявляется на первом году жизни.
- Повышенная возбудимость.
- Задержка развития.
- Обильное слюнотечение.
- Сниженное артериальное давление.
- Частое повышение температуры тела.
- Сухожильная гиперрефлексия (повышение рефлексов) или спастический тетрапарез (обессиливание всех четырех конечностей).
- Миоклоническая эпилепсия (генерализованные приступы, преимущественно возникающие после пробуждения).
- Микроцефалия.
Отличительная особенность второго типа ― гибель нейронов, нарушение метаболизма фолатов, а также кальцификация в различных отделах головного мозга. Болезнь быстро прогрессирует, может привести к смерти ребенка в течение 2 — 3 лет.
III тип. Симптомы дефицита пирувоилтетрагидроптеринсинтетазы схож с проявлениями болезни Паркинсона:
- Постуральная нестабильность и трудности походки. Сложно либо невозможно поддерживать определенное положение всего тела или конечностей.
- Гипокинезия (низкая двигательная активность, ограниченный темп и объем движений).
- Гиперсаливация (повышенное слюноотделение).
- Нарушения глотания.
- Окулогирные кризы (симметричное отклонение обоих глаз, обычно направленное вверх).
В 80% случаев этот тип заболевания сопровождается снижением количества биогенных аминов в ликворе. Лечение затруднено тем, что раннее снижение концентрации ФА может вызвать серьезные патологические изменения. Несоблюдение диетотерапии приведет к замедлению развития речи, низкому интеллекту, проблемам с памятью.
Диагностика
Выявить фенилкетонурию можно в первые дни после рождения до появления какой-либо симптоматики. Для определение концентрации фениламина в крови проводят:
- микробиологический тест;
- хроматографию;
- флюориметрию;
- масс-спектрометрию.
Во всех случаях биологическим материалом выступают сухие пятна капиллярной крови младенца.
С недавнего времени анализ на фенилкетонурию входит в программу неонатального скрининга. Его проводят всем новорожденным, особенно важно исследование для недоношенных детей. Критерий диагностирования ― повышенная концентрация фенилаланина при норме 0 — 2 мг/дл. Повышенный уровень требует проведения уточняющей диагностики. Потребуется установить сам факт наличия фенилкетонурии и выявить ее причину.
- Если скрининг-тест показал высокие результаты уровня ФА, дополнительно может быть назначено:
- Фенилаланин-нагрузочная диагностика для выявления нозологической формы заболевания.
- Молекулярно-генетический анализ для установления формы: классическая, II или III тип.
- Секвенирование гена РАН, если молекулярно-генетическая диагностика дала отрицательный результат по гену ФАГ.
- Анализ на птерины в урине для исключения птерин-зависимых форм.
Дифференциальное диагностирование фенилкетонурии проводят с такими патологиями, как нарушение функции печени, галактоземия и с другими заболеваниями.
Лечение
Симптоматическая терапия при любой формой фенилкетонурии неэффективна. Существует только один способ предотвратить негативные последствия заболевания ― диетотерапия. Из рациона исключают высокобелковые и содержащие фенилаланин продукты. Недостающее количество белка восполняют специализированным лечебным питанием, с максимально низким содержанием аминокислоты ФА или полностью ее лишенным. Следует учитывать, что эффективность терапии напрямую зависит от времени начала коррекции и уже произошедших патологических изменений.
Цель лечебного питания при классической форме заболевания ― это предотвращение развития нарушений ЦНС, физического и умственного развития. Легкая форма ГФА допускает расширение диеты под строгим наблюдением врача за состоянием ребенка и биохимическими показателями. Под запретом: мясо, рыба, орехи, шоколад и бобовые, все виды яиц, творог и сыры. Также следует исключить продукты, содержащие искусственный подсластитель аспартам.
Критерий эффективности лечения ― уровень ФА в крови.
Наследование аутосомно-рецессивной фенилкетонутии
В том случае, если будущая мама заболела фенилкетонурией, риск унаследования заболевания ребенком зависит от генотипа будущего отца. Если отец ребенка не является носителем аномального гена, ребенок родится здоровым (но будет носителем). С другой стороны, если отец ребенка несет ген фенилкетонурии, вероятность рождения больного ребенка составляет 50%.
Беременным также очень важно соблюдать диету — слишком высокий уровень фенилаланина может привести к порокам у ребенка (порокам сердца, конечностей или умственной отсталости).
Прогноз и профилактика
Проведения массового скрининга в родильных домах позволяет своевременно выявить генетическое отклонение. Вовремя начать соблюдение диетотерапии и, как следствие, предотвратить тяжелые последствия. В противном случае прогноз в отношении умственного развития неблагоприятный.
Классическая ФКУ имеет благоприятный прогноз если диагностирована в первые недели жизни ребенка и соблюдаются все требования врачей. Такие дети ходят в обычные школы, способны получить высшее образовании и вести нормальный образ жизни.
Во время подготовки к беременности пара должна пройти предварительное генетическое тестирование на наличие мутаций в гене РАН. Если у одного из родителей есть дефектный ген, шанс родить ребенка с ФКУ 1:4 и 100% если оба родителя больны.
Женщины с установленной фенилкетонурией при беременности и грудном вскармливании должны соблюдать строгую диету. Высокая концентрация аминокислоты в крови и околоплодных водах оказывает серьезное тератогенное воздействие на плод.
При беременности
Женщинам с фенилкетонурией, которых успешно лечили с помощью диеты, следует крайне внимательно относится к своему состоянию во время вынашивания ребенка, ведь даже при генетически нормальном развитии плода высок риск нарушений из-за повышенного уровня фенилаланина и его метаболитов в кровоток матери как серьезного тератогенного фактора с нейротропной токсичностью. Поэтому, перед зачатием таким пациенткам рекомендуют вернуться к строгой диете и придерживаться ее всю беременность.
Особая диета должна ограничивать потребление до 15-20 мг/кг фенилаланина в сутки, и при этом следить, чтобы не развивался дефицит незаменимых аминокислот.









{kind=link}
{kind=link}
{kind=link}
{kind=link}