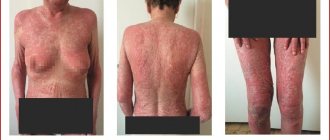
Предпосылки к проведению исследования
Повышенная концентрация холестерина (ХС) липопротеинов низкой плотности (ЛПНП) относится к известным и модифицируемым факторам риска развития осложнений сердечно-сосудистых заболеваний (ССЗ). Моноклональные антитела, ингибирующие пропротеинконвертазу субтилизина/кексина 9-го типа (proprotein convertase subtilisin/kexin 9 — PCSK9), представляют собой новый класс препаратов, которые эффективно снижают уровень ХС ЛПНП крови [1]. Эволокумаб относится к такому классу препаратов и представляет собой полностью моноклинальные антитела человека; применение этого препарата приводит к снижению уровня ХС ЛПНП примерно на 60% [2—6].
Результаты генетических исследований свидетельствовали о связи между наличием мутации потери функции гена PCSK9
с более низким уровнем ХС ЛПНП и снижением риска развития инфаркта миокарда (ИМ) [7, 8]. Более того, результаты поискового анализа данных, полученных при длительном наблюдении за участниками исследований II и III фазы по оценке эффективности применения ингибиторов PCSK9, свидетельствовали о статистически значимом снижении риска развития осложнений ССЗ [9, 10]. Однако в ходе выполнения таких исследований развилось в целом лишь ненамного более 100 неблагоприятных исходов.
Репата, 1 шт., 1 мл, 140 мг/мл, раствор для подкожного введения
Механизм действия Эволокумаб является полностью человеческим моноклональным иммуноглобулином G2 (IgG2), ингибирующим пропротеин конвертазу субтилизин/кексин типа 9 (PCSK9). Эволокумаб селективно и с высокой степенью аффинности связывается с PCSK9 и ингибирует связывание циркулирующей PCSK9 с рецептором липопротеинов низкой плотности (Р-ЛПНП) на поверхности клеток печени, таким образом предотвращая PCSK9-опосредованный распад Р-ЛПНП. Как результат, повышение экспрессии Р-ЛПНП в печени приводит к снижению сывороточной концентрации холестерина липопротеинов низкой плотности (ХС-ЛПНП). Фармакодинамические свойства Было показано, что у пациентов с первичной гиперлипидемией и смешанной дислипидемией эволокумаб снижает концентрации несвязанной PCSK9, ХС-ЛПНП, общего холестерина (ОХ), аполипопротеина B (АпоВ), холестерина липопротеинов невысокой плотности (ХС не-ЛПВП), холестерина липопротеинов очень низкой плотности (ХС-ЛПОНП), триглицеридов и липопротеина(а) (Лп[а]), повышает концентрации холестерина липопротеинов высокой плотности\0009(Хс-ЛПВП) и аполипопротеина A1(АпоА1), улучшая соотношение ОС/Хс-ЛПВП, АпоВ/аполипопротеин А1 (АпоА1). Однократное подкожное введение 140 или 420 мг эволокумаба приводит к максимальной супрессии цикрулирующей несвязанной PCSK9 через 4 часа, что сопровождается снижением ХС-ЛПНП, достигающего среднего надира к 14 и 21 дню, соответственно. Изменения концентрации несвязанной PCSK9 и сывороточных липопротеинов обратимы после отмены эволокумаба. Не отмечено компенсаторного увеличения продукции PCSK9 и ХС-ЛПНП во время лечения, равно как и после выведения эволокумаба не отмечено повышения концентраций несвязанной PCSK9 или ХС-ЛПНП (нет «синдрома рикошета»). При режиме дозирования 140 мг эволокумаба каждые две недели или 420 мг эволокумаба раз в месяц максимальное снижение ХС-ЛПНП достигало от -72% до -57% от начальных значений по сравнению с плацебо. Режимы дозирования эквивалентны в отношении среднего снижения ХС-ЛПНП (среднее на неделях 10 и 12). Аналогичное снижение ХС-ЛПНП наблюдалось как при применении эволокумаба в монотерапии, так и в составе комбинированной терапии с другими гиполипидемическими препаратами. Эффект в отношении снижения ХС-ЛПНП стабилен, максимальная продолжительность терапии в настоящий момент составляет 112 недель. Внешние и внутренние факторы, такие как демографические характеристики, одновременно применяемая терапия, вариабельность лабораторных показателей и статус заболевания не влияют на ответ на терапию эволокумабом (см. раздел «Режим дозирования»). Иммуногенность Как и в случае любых других терапевтических протеинов, существует потенциальный риск развития иммуногенности.Иммуногенность определяли с помощью иммунохемилюминисцентного связывания с целью обнаружения антител к эволокумабу. В случае выявления у пациентов при иммунологическом скрининге антител к эволокумабу дополнительно проводили биологический анализ для оценки того, являются ли эти антитела нейтрализующими. В клинических исследованиях у 0.1% пациентов (7 из 4846 пациентов с первичной гиперхолестеринемией и смешанной дислипидемией и ни у одного из 80 пациентов с гомозиготной семейной гиперхолестеринемией [ГоСГХС]), получивших как минимум 1 дозу эволокумаба, обнаружены связывающие антитела (у 4 пациентов — транзиторные антитела). Для этих пациентов проводили дополнительный анализ на нейтрализующие антитела. Нейтрализующих антител не обнаружено ни у одного из пациентов. Обнаруженные связывающие антитела не приводили к изменению фармакокинетических параметров препарата, не влияли на терапевтический ответ или безопасность препарата. Клиническая эффективность и безопасность Результаты клинических исследований эволокумаба доказывают, что ингибирование активности PCSK9 эволокумабом обеспечивает снижение концентрации ХС-ЛПНП в сыворотке и улучшение других показателей липидного обмена. Данные результаты демонстрируют стабильную эффективность эволокумаба в отношении улучшения показателей липидного обмена у пациентов с первичной гиперлипидемией (гетерозиготной семейной и несемейной) и смешанной дислипидемией и ГоСГХС во всех популяциях и с любым дизайном исследований. Режимы дозирования 140 мг эволокумаба раз в две недели (Q2W) и 420 мг (QM) эволокумаба раз в месяц являются клинически эквивалентными у пациентов с первичной гиперхолестеринемией и смешанной дислипидемией в отношении снижения ХС-ЛПНП, общего холестерина, АпоB, ХС не-ЛПВП, ХС-ЛПОНП, триглицеридов и Лп(а); повышения концентрации ХС-ЛПВП и АпоA1 и улучшения соотношения общий холестерин/ХС-ЛПВП, АпоB/АпоA1. В результате терапии эволокумабом было достигнуто снижение концентрации ХС-ЛПНП приблизительно на 55-75%, которое сохранялось на протяжении всего периода проведения долгосрочной терапии. Максимальный ответ достигался, как правило, через 12 недели после введения 140 мг раз Q2W и 420 мг QM, соответственно. У 80-85% пациентов, получавших эволокумаб в любой дозировке, наблюдалось снижение концентрации ХС-ЛПНП на более чем 50% в среднем к 10 — 12 неделям применения. Эволокумаб превосходил эзетимиб в отношении снижения концентрации ХС-ЛПНП, Применение эволокумаба 140 мг Q2W и 420 мг QM было эффективным во всех подгруппах, относящихся к плацебо и эзетимибу, при этом каких-либо значимых различий между подгруппами, определенными такими характеристиками пациентов как возраст, расовая принадлежность, пол, регион происхождения, индекс массы тела (ИМТ), степень риска по Национальной образовательной программе по холестерину (NCEP), доза и интенсивность статинов, статус курения, исходный риск развития ишемической болезни сердца (ИБС), ранняя ИБС в семейном анамнезе, переносимость или непереносимость глюкозы (т.е. сахарный диабет 2 типа, метаболический синдром или ни то, ни другое), артериальная гипертензия, исходная несвязанная PCSK9, исходная концентрация ХС-ЛПНП и исходная концентрация триглицеридов, не наблюдалось. Результаты общего анализа эффективности в исследованиях ГоСГХС свидетельствуют о том, что эволокумаб эффективно снижал концентрации ХС-ЛПНП, общего холестерина, АпоB и ХС-неЛПВП у пациентов с ГоСГХС. При долгосрочном лечении эволокумабом в дозах 420 мг QM и 420 мг Q2W наблюдался продолжительный терапевтический эффект, что подтверждается снижением концентрации ХС-ЛПНП приблизительно на 20% — 30% у пациентов c ГоСГХС, не получавших лечения аферезом, и приблизительно у 15% — 25% пациентов с ГоСГХС, получавших аферез. В целом, каких-либо различий в отношении безопасности или эффективности эволокумаба между группами в возрасте 12 лет и старше и взрослых пациентов с ГоСГХС не наблюдалось. Соотношение пациентов с нежелательными явлениями было в целом сбалансированным между группами в течение всех 3 периодов сбора комплексного массива данных по безопасности, а также между подгруппами и схемами лечения. Не отмечалось каких-либо связанных с безопасностью проблем в отношении нежелательных явлений, сообщавшихся для других видов гиполипидемической терапии (т.е. эпизодов сахарного диабета, а также осложнений со стороны печени и мышечной ткани), а также явлений, которые теоретически могут быть сопряжены с ингибированием PCSK9/повышением экспрессии рецепторов ЛПНП (т.е. эпизоды развития гепатита С). Каких-либо признаков, свидетельствующих о риске развития нейрокогнитивных осложнений при применении эволокумаба, не наблюдалось. Нейрокогнитивные осложнения в исследованиях с контролем плацебо или активными контролями были сходными. Виды и количество нежелательных явлений во всех исследованиях были сопоставимыми, как при применении эволокумаба в качестве монотерапии, так и в составе комбинированной терапии (со статинами в сочетании с эзетимибом или без него) или у пациентов с непереносимостью статинов. Не было выявлено новых сигналов, касающихся безопасности препарата, при сравнительном изучении данных о нежелательных явлениях, развившихся в ходе исследований пациентов с ГоСГХС и данных о нежелательных явлениях, развившихся в исследованиях первичной гиперлипидемии (гетерозиготной семейной и несемейной) и смешанной дислипидемии.
Фармакокинетика
Фармакокинетика эволокумаба после подкожного введения демонстрирует нелинейный характер. Всасывание Медианная максимальная сывороточная концентрация достигалась в течение 3 — 4 дней с расчетной абсолютной биодоступностью 72% после однократной подкожной инъекции эволокумаба 140 мг или 420 мг здоровым добровольцам. Среднее значение максимальной концентрации (Cmax mean (SD)) составило 18.6 (7.3) мкг/мл после введения дозы 140 мг. Конечная площадь под кривой «концентрация-время» (AUClast) составила 188 (98.6) сутки»мкг/мл. Аналогичные значения Cmax и AUClast составили 59.0 (17.2) мкг/мл и 924 (346) сутки»мкг/мл, соответственно, после введения дозы 420 мг. Распределение Средний расчетный объем распределения в равновесном состоянии составил 3.3 (0.5) л после введения однократной дозы 420 мг эволокумаба внутривенно, что предполагает ограниченное распределение эволокумаба в ткани. Метаболизм Рассчитанное среднее системного клиренса составило 12 (2) мл/ч после внутривенного введения однократно 420 мг эволокумаба. Повторное подкожное введение эволокумаба на протяжении 12 недель в клинических исследованиях приводило к дозопропорциональному увеличению экспозиции для режимов дозирования 140 мг и больше. Приблизительно двух- и трехкратная кумуляция наблюдалась при минимальной сывороточной концентрации (Cmin [SD]) 7.21 [6.6]) при режимах дозирования 140 мг каждые 2 недели или 420 мг раз в месяц (Cmin [SD] 11.2 [10.8]). Минимальная сывороточная концентрация достигала состояния равновесия к 12 неделе дозирования. Расчетный эффективный период полувыведения эволокумаба составил от 11 до 17 дней. Не было отмечено изменений сывороточных концентраций эволокумаба в течение 124 недель, связанных со временем. Выведение Поскольку эволокумаб представляет собой полностью человеческое моноклональное антитело IgG2, клиренс эволокумаба обусловлен специфическим связыванием и образованием комплекса с целевым лигандом, PCSK9, равно как и стандартными путями клиренса IgG в ретикуло-эндотелиальной системе. Эволокумаб распадается до малых пептидов и аминокислот посредством этих путей катаболизма. Увеличение клиренса приблизительно на 20% отмечено при комбинированном применении со статинами. Это увеличение частично обусловлено вызванным статинами увеличением концентраций PCSK9 и не оказывает негативного эффекта на фармакодинамику эволокумаба в отношении липидов. Фармакокинетический анализ в популяциях не выявил значимых различий в сывороточных концентрациях эволокумаба у пациентов с гиперхолестеринемией (семейной и несемейной), при одновременном приеме статинов. Отдельные группы пациентов Согласно результатам фармакокинетического анализа в популяциях не требуется коррекции дозы в зависимости от возраста, расы или пола. Масса тела влияет на фармакокинетику эволокумаба, однако значимо не влияет на гиполипидемический эффект эволокумаба. Следовательно, коррекции режима дозирования в зависимости от массы тела не требуется. Фармакокинетический анализ в популяциях по объединенным данным клинических исследований не выявил различий в фармакокинетике препарата у пациентов с нарушениями функции почек слабой и умеренной степени по сравнению с пациентами с нормальной функцией почек. Препарат вводили в виде одной подкожной инъекции 140 мг 8 пациентам со слабой степенью нарушения функции печени, 8 пациентам с умеренной степенью нарушения и 8 здоровым добровольцам. Экспозиция к эволокумабу снижалась на 40% — 50% по сравнению со здоровыми добровольцами. Тем не менее, базовые концентрации PCSK9, равно как и степень и время нейтрализации PCSK9 оставались схожими во всех трех группах. Таким образом, отмечался сходный эффект на снижение ХС-ЛПНП.
Больные
В исследование включали больных 40—85 лет с клиническими проявлениями ССЗ, обусловленного атеросклерозом, которое диагностировали при наличии в анамнезе ИМ, негеморрагического инсульта или заболевания периферических артерий с клиническими проявлениями, у которых имелись дополнительные характеристики, свидетельствующие о более высоком риске развития осложнений ССЗ. Кроме того, для включения в исследование концентрация ХС ЛПНП должна была достигать 1,8 ммоль/л и более или концентрация холестерина липопротеинов всех типов, кроме липопротеинов высокой плотности (ХС не-ЛПВП) 2,6 ммоль/л и более, несмотря на применение оптимизированного режима терапии, который должен был включать преимущественно интенсивный режим приема статина с использованием хотя бы 20 мг аторвастатина или эквивалентной дозы другого статина в сочетании с эзетимибом или в его отсутствие. Подробно исходные характеристики больных, включенных в исследование, представлены в таблице.
Таблица. Исходные характеристики больных, включенных в исследование* Примечание.* — данные представлены как среднее ± стандартное отклонение или как медиана значений (межквартильный диапазон), если не указано другое. ** — формально отсутствовали статистически значимые различия между группами по исходным характеристикам за исключением различий по массе тела (p=0,01) и частоте применения аспирина, ингибитора P2Y12 или их сочетанного использования (p=0,03). ССЗ — сердечно-сосудистое заболевание; ИМ — инфаркт миокарда; АПФ — ангиотензинпревращающий фермент; БРА — блокатор рецепторов ангиотензина II; ХС — холестерин; ЛПНП — липопротеины низкой плотности; ЛПВП — липопротеины высокой плотности.
Репата, 140 мг/мл, раствор для подкожного введения, 1 мл, 1 шт.
Механизм действия Эволокумаб является полностью человеческим моноклональным иммуноглобулином G2 (IgG2), ингибирующим пропротеин конвертазу субтилизин/кексин типа 9 (PCSK9). Эволокумаб селективно и с высокой степенью аффинности связывается с PCSK9 и ингибирует связывание циркулирующей PCSK9 с рецептором липопротеинов низкой плотности (Р-ЛПНП) на поверхности клеток печени, таким образом предотвращая PCSK9-опосредованный распад Р-ЛПНП. Как результат, повышение экспрессии Р-ЛПНП в печени приводит к снижению сывороточной концентрации холестерина липопротеинов низкой плотности (ХС-ЛПНП). Фармакодинамические свойства Было показано, что у пациентов с первичной гиперлипидемией и смешанной дислипидемией эволокумаб снижает концентрации несвязанной PCSK9, ХС-ЛПНП, общего холестерина (ОХ), аполипопротеина B (АпоВ), холестерина липопротеинов невысокой плотности (ХС не-ЛПВП), холестерина липопротеинов очень низкой плотности (ХС-ЛПОНП), триглицеридов и липопротеина(а) (Лп[а]), повышает концентрации холестерина липопротеинов высокой плотности\0009(Хс-ЛПВП) и аполипопротеина A1(АпоА1), улучшая соотношение ОС/Хс-ЛПВП, АпоВ/аполипопротеин А1 (АпоА1). Однократное подкожное введение 140 или 420 мг эволокумаба приводит к максимальной супрессии цикрулирующей несвязанной PCSK9 через 4 часа, что сопровождается снижением ХС-ЛПНП, достигающего среднего надира к 14 и 21 дню, соответственно. Изменения концентрации несвязанной PCSK9 и сывороточных липопротеинов обратимы после отмены эволокумаба. Не отмечено компенсаторного увеличения продукции PCSK9 и ХС-ЛПНП во время лечения, равно как и после выведения эволокумаба не отмечено повышения концентраций несвязанной PCSK9 или ХС-ЛПНП (нет «синдрома рикошета»). При режиме дозирования 140 мг эволокумаба каждые две недели или 420 мг эволокумаба раз в месяц максимальное снижение ХС-ЛПНП достигало от -72% до -57% от начальных значений по сравнению с плацебо. Режимы дозирования эквивалентны в отношении среднего снижения ХС-ЛПНП (среднее на неделях 10 и 12). Аналогичное снижение ХС-ЛПНП наблюдалось как при применении эволокумаба в монотерапии, так и в составе комбинированной терапии с другими гиполипидемическими препаратами. Эффект в отношении снижения ХС-ЛПНП стабилен, максимальная продолжительность терапии в настоящий момент составляет 112 недель. Внешние и внутренние факторы, такие как демографические характеристики, одновременно применяемая терапия, вариабельность лабораторных показателей и статус заболевания не влияют на ответ на терапию эволокумабом (см. раздел «Режим дозирования»). Иммуногенность Как и в случае любых других терапевтических протеинов, существует потенциальный риск развития иммуногенности.Иммуногенность определяли с помощью иммунохемилюминисцентного связывания с целью обнаружения антител к эволокумабу. В случае выявления у пациентов при иммунологическом скрининге антител к эволокумабу дополнительно проводили биологический анализ для оценки того, являются ли эти антитела нейтрализующими. В клинических исследованиях у 0.1% пациентов (7 из 4846 пациентов с первичной гиперхолестеринемией и смешанной дислипидемией и ни у одного из 80 пациентов с гомозиготной семейной гиперхолестеринемией [ГоСГХС]), получивших как минимум 1 дозу эволокумаба, обнаружены связывающие антитела (у 4 пациентов — транзиторные антитела). Для этих пациентов проводили дополнительный анализ на нейтрализующие антитела. Нейтрализующих антител не обнаружено ни у одного из пациентов. Обнаруженные связывающие антитела не приводили к изменению фармакокинетических параметров препарата, не влияли на терапевтический ответ или безопасность препарата. Клиническая эффективность и безопасность Результаты клинических исследований эволокумаба доказывают, что ингибирование активности PCSK9 эволокумабом обеспечивает снижение концентрации ХС-ЛПНП в сыворотке и улучшение других показателей липидного обмена. Данные результаты демонстрируют стабильную эффективность эволокумаба в отношении улучшения показателей липидного обмена у пациентов с первичной гиперлипидемией (гетерозиготной семейной и несемейной) и смешанной дислипидемией и ГоСГХС во всех популяциях и с любым дизайном исследований. Режимы дозирования 140 мг эволокумаба раз в две недели (Q2W) и 420 мг (QM) эволокумаба раз в месяц являются клинически эквивалентными у пациентов с первичной гиперхолестеринемией и смешанной дислипидемией в отношении снижения ХС-ЛПНП, общего холестерина, АпоB, ХС не-ЛПВП, ХС-ЛПОНП, триглицеридов и Лп(а); повышения концентрации ХС-ЛПВП и АпоA1 и улучшения соотношения общий холестерин/ХС-ЛПВП, АпоB/АпоA1. В результате терапии эволокумабом было достигнуто снижение концентрации ХС-ЛПНП приблизительно на 55-75%, которое сохранялось на протяжении всего периода проведения долгосрочной терапии. Максимальный ответ достигался, как правило, через 12 недели после введения 140 мг раз Q2W и 420 мг QM, соответственно. У 80-85% пациентов, получавших эволокумаб в любой дозировке, наблюдалось снижение концентрации ХС-ЛПНП на более чем 50% в среднем к 10 — 12 неделям применения. Эволокумаб превосходил эзетимиб в отношении снижения концентрации ХС-ЛПНП, Применение эволокумаба 140 мг Q2W и 420 мг QM было эффективным во всех подгруппах, относящихся к плацебо и эзетимибу, при этом каких-либо значимых различий между подгруппами, определенными такими характеристиками пациентов как возраст, расовая принадлежность, пол, регион происхождения, индекс массы тела (ИМТ), степень риска по Национальной образовательной программе по холестерину (NCEP), доза и интенсивность статинов, статус курения, исходный риск развития ишемической болезни сердца (ИБС), ранняя ИБС в семейном анамнезе, переносимость или непереносимость глюкозы (т.е. сахарный диабет 2 типа, метаболический синдром или ни то, ни другое), артериальная гипертензия, исходная несвязанная PCSK9, исходная концентрация ХС-ЛПНП и исходная концентрация триглицеридов, не наблюдалось. Результаты общего анализа эффективности в исследованиях ГоСГХС свидетельствуют о том, что эволокумаб эффективно снижал концентрации ХС-ЛПНП, общего холестерина, АпоB и ХС-неЛПВП у пациентов с ГоСГХС. При долгосрочном лечении эволокумабом в дозах 420 мг QM и 420 мг Q2W наблюдался продолжительный терапевтический эффект, что подтверждается снижением концентрации ХС-ЛПНП приблизительно на 20% — 30% у пациентов c ГоСГХС, не получавших лечения аферезом, и приблизительно у 15% — 25% пациентов с ГоСГХС, получавших аферез. В целом, каких-либо различий в отношении безопасности или эффективности эволокумаба между группами в возрасте 12 лет и старше и взрослых пациентов с ГоСГХС не наблюдалось. Соотношение пациентов с нежелательными явлениями было в целом сбалансированным между группами в течение всех 3 периодов сбора комплексного массива данных по безопасности, а также между подгруппами и схемами лечения. Не отмечалось каких-либо связанных с безопасностью проблем в отношении нежелательных явлений, сообщавшихся для других видов гиполипидемической терапии (т.е. эпизодов сахарного диабета, а также осложнений со стороны печени и мышечной ткани), а также явлений, которые теоретически могут быть сопряжены с ингибированием PCSK9/повышением экспрессии рецепторов ЛПНП (т.е. эпизоды развития гепатита С). Каких-либо признаков, свидетельствующих о риске развития нейрокогнитивных осложнений при применении эволокумаба, не наблюдалось. Нейрокогнитивные осложнения в исследованиях с контролем плацебо или активными контролями были сходными. Виды и количество нежелательных явлений во всех исследованиях были сопоставимыми, как при применении эволокумаба в качестве монотерапии, так и в составе комбинированной терапии (со статинами в сочетании с эзетимибом или без него) или у пациентов с непереносимостью статинов. Не было выявлено новых сигналов, касающихся безопасности препарата, при сравнительном изучении данных о нежелательных явлениях, развившихся в ходе исследований пациентов с ГоСГХС и данных о нежелательных явлениях, развившихся в исследованиях первичной гиперлипидемии (гетерозиготной семейной и несемейной) и смешанной дислипидемии.
Фармакокинетика
Фармакокинетика эволокумаба после подкожного введения демонстрирует нелинейный характер. Всасывание Медианная максимальная сывороточная концентрация достигалась в течение 3 — 4 дней с расчетной абсолютной биодоступностью 72% после однократной подкожной инъекции эволокумаба 140 мг или 420 мг здоровым добровольцам. Среднее значение максимальной концентрации (Cmax mean (SD)) составило 18.6 (7.3) мкг/мл после введения дозы 140 мг. Конечная площадь под кривой «концентрация-время» (AUClast) составила 188 (98.6) сутки»мкг/мл. Аналогичные значения Cmax и AUClast составили 59.0 (17.2) мкг/мл и 924 (346) сутки»мкг/мл, соответственно, после введения дозы 420 мг. Распределение Средний расчетный объем распределения в равновесном состоянии составил 3.3 (0.5) л после введения однократной дозы 420 мг эволокумаба внутривенно, что предполагает ограниченное распределение эволокумаба в ткани. Метаболизм Рассчитанное среднее системного клиренса составило 12 (2) мл/ч после внутривенного введения однократно 420 мг эволокумаба. Повторное подкожное введение эволокумаба на протяжении 12 недель в клинических исследованиях приводило к дозопропорциональному увеличению экспозиции для режимов дозирования 140 мг и больше. Приблизительно двух- и трехкратная кумуляция наблюдалась при минимальной сывороточной концентрации (Cmin [SD]) 7.21 [6.6]) при режимах дозирования 140 мг каждые 2 недели или 420 мг раз в месяц (Cmin [SD] 11.2 [10.8]). Минимальная сывороточная концентрация достигала состояния равновесия к 12 неделе дозирования. Расчетный эффективный период полувыведения эволокумаба составил от 11 до 17 дней. Не было отмечено изменений сывороточных концентраций эволокумаба в течение 124 недель, связанных со временем. Выведение Поскольку эволокумаб представляет собой полностью человеческое моноклональное антитело IgG2, клиренс эволокумаба обусловлен специфическим связыванием и образованием комплекса с целевым лигандом, PCSK9, равно как и стандартными путями клиренса IgG в ретикуло-эндотелиальной системе. Эволокумаб распадается до малых пептидов и аминокислот посредством этих путей катаболизма. Увеличение клиренса приблизительно на 20% отмечено при комбинированном применении со статинами. Это увеличение частично обусловлено вызванным статинами увеличением концентраций PCSK9 и не оказывает негативного эффекта на фармакодинамику эволокумаба в отношении липидов. Фармакокинетический анализ в популяциях не выявил значимых различий в сывороточных концентрациях эволокумаба у пациентов с гиперхолестеринемией (семейной и несемейной), при одновременном приеме статинов. Отдельные группы пациентов Согласно результатам фармакокинетического анализа в популяциях не требуется коррекции дозы в зависимости от возраста, расы или пола. Масса тела влияет на фармакокинетику эволокумаба, однако значимо не влияет на гиполипидемический эффект эволокумаба. Следовательно, коррекции режима дозирования в зависимости от массы тела не требуется. Фармакокинетический анализ в популяциях по объединенным данным клинических исследований не выявил различий в фармакокинетике препарата у пациентов с нарушениями функции почек слабой и умеренной степени по сравнению с пациентами с нормальной функцией почек. Препарат вводили в виде одной подкожной инъекции 140 мг 8 пациентам со слабой степенью нарушения функции печени, 8 пациентам с умеренной степенью нарушения и 8 здоровым добровольцам. Экспозиция к эволокумабу снижалась на 40% — 50% по сравнению со здоровыми добровольцами. Тем не менее, базовые концентрации PCSK9, равно как и степень и время нейтрализации PCSK9 оставались схожими во всех трех группах. Таким образом, отмечался сходный эффект на снижение ХС-ЛПНП.
Вмешательство
Больных в соотношении 1:1 распределяли в группу подкожного введения эволокумаба (либо по 140 мг каждые 2 нед, либо по 420 мг 1 раз в месяц в зависимости от предпочтений больного) и группу плацебо. Рандомизация выполнялась с использованием двойного слепого метода и централизованной компьютеризированной системы со стратификацией больных в зависимости от результатов последнего измерения концентрации ХС ЛПНП в период предварительного обследования (менее 2,2 ммоль/л или 2,2 ммоль/л и более) и от региона проживания.
Критерии оценки/Клинические исходы
Основной комбинированный показатель эффективности: частота развития таких тяжелых осложнений ССЗ, как:
— смерть от осложнений ССЗ
— инсульт
— госпитализация по поводу нестабильной стенокардии
— выполнение реваскуляризации миокарда.
Главный дополнительный комбинированный показатель эффективности частоты развития таких неблагоприятных исходов, как:
— смерть от осложнений ССЗ
— инсульт.
Безопасность исследуемой терапии оценивали с помощью получения данных о развитии нежелательных явлений (НЯ) и результатов анализов, выполненных в центральной лаборатории.
Члены центрального комитета по подтверждению неблагоприятных исходов, работой которого руководила исследовательская группа TIMI, рассматривали все возможные неблагоприятные исходы, включенные в показатели эффективности, а также случаи развития сахарного диабета (СД) в отсутствие информации о результатах распределения больных в группу определенной тактики и концентрации липидов в крови.
Методы статистического анализа
Анализ основного показателя основывался на оценке продолжительности периода между рандомизацией и развитием первого неблагоприятного исхода, включенного в основной комбинированный показатель эффективности. В случае статистически значимого снижения основного комбинированного показателя в группе эволокумаба по сравнению с группой плацебо (p<0,05) с использованием иерархического подхода переходили к проверке влияния применения эволокумаба на главный дополнительный показатель, а затем на смертность от осложнений ССЗ при уровне статистической значимости 0,05.
Все виды статистического анализа эффективности выполняли исходя из допущения, что у всех больных применялось назначенное лечение. Для пропущенных данных о развитии неблагоприятных клинических исходов подстановку не выполняли. В анализ безопасности включали данные о всех больных, которые были рандомизированы и которым ввели хотя бы одну дозу исследуемого препарата в случае, если для них были доступны данные, полученные после применения такой дозы.
Расчет объема выборки основывался на частоте развития неблагоприятных клинических исходов, включенных в главный дополнительный показатель; причем было установлено, что для достижения 90% статистической мощности исследования в целях выявления различий между группой эволокумаба и группой плацебо по такому показателю в 15% требовалось развитие 1630 указанных исходов. Отношения рисков и 95% ДИ рассчитывали с помощью модели пропорциональных рисков Кокса с использованием стратификационных факторов в качестве ковариат, а значение p рассчитывали в ходе выполнения анализа продолжительности периода до развития неблагоприятного клинического исхода с помощью лог-ранговых критериев.
Результаты
С января 2013 г. по июнь 2015 г. в целом в исследование были включены 27 564 больных: в группу эволокумаба и группу плацебо 13 784 и 13 780 больных соответственно. Не отмечалось существенных различий между группами по основным исходным характеристикам больных (см. таблицу). Средний возраст больных составлял 63 года; 24,6% женщин; 81,1% больных ранее переносили ИМ, 19,4% — негеморрагический инсульт и у 13,2% имелись клинические проявления заболевания периферических артерий. При включении в исследование интенсивный и умеренно интенсивный режим терапии статинами (в соответствии с объединенными рекомендациями Американской коллегии кардиологов и Американской ассоциации кардиологов [11]) применялся у 69,3 и 30,4% больных соответственно; кроме того, 5,2% больных также принимали эзетимиб. В ходе выполнения исследования только у 9,8% больных изменялась базовая гиполипидемическая терапия. Частота использования терапии, направленной на вторичную профилактику осложнений ССЗ, в целом была высокой: при включении в исследование антиагреганты, β-блокаторы, ингибиторы ангиотензинпревращающего фермента (АПФ) или блокаторы рецепторов ангиотензина II, антагонисты альдостерона или их сочетание применяли 92,3, 75,6 и 78,2% больных соответственно.
В целом 27 525, или 99,9%, больных была введена хотя бы 1 доза исследуемого препарата. Стойко прекратили применение исследуемого препарата 12,5% больных (5,7% в год), а отказались от продолжения участия в исследовании 0,7% больных (0,3% в год) и с менее 0,1% больных (0,03% в год) был потерян контакт. Причем такая частота была сходной в обеих группах. Медиана продолжительности наблюдения достигала 26 мес (межквартильный диапазон — МКД от 22 до 30 мес), что соответствовало общему объему наблюдения 59 865 человеко-лет. Оценка клинических исходов, включенных в основной показатель, была выполнена для 99,5% возможных человеко-лет наблюдения.
Медиана концентрации ХС ЛПНП при включении в исследование в целом составляла 2,4 ммоль/л (МКД от 2,1 до 2,8 ммоль/л). Через 48 нед после рандомизации снижение концентрации ХС ЛПНП в процентах, которую рассчитывали с помощью метода наименьших квадратов, в группе эволокумаба по сравнению с группой плацебо достигало 59% (при 95% ДИ от 58 до 60%; p<0,001) при среднем абсолютном снижении уровня ХС ЛПНП в крови на 1,45 ммоль/л (при 95% ДИ от 1,43 до 1,47) до концентрации ХС ЛПНП, медиана которой составляла 0,78 ммоль/л (МКД от 0,49 до 1,2). Степень снижения концентрации ХС ЛПНП сохранялась в ходе выполнения исследования. Через 48 нед в группе эволокумаба уровень ХС ЛПНП снижался до 1,8 ммоль/л или менее, до 1 ммоль/л или менее и до 0,65 ммоль/л или менее у 87, 67 и 42% больных соответственно, в то время как в группе плацебо снижение концентрации ХС ЛПНП до такого уровня отмечалось у 18, 0,5 и менее 0,1% больных соответственно (p<0,001 для всех сравнений между группой эволокумаба и группой плацебо). Точно так же применение эволокумаба по сравнению с плацебо приводило к соответствующему снижению уровня других атерогенных липидов: через 48 нед в группе эволокумаба по сравнению с группой плацебо концентрация ХС не-ЛПВП и аполипопротеина В снижалась на 52 и 49% соответственно (p<0,001 для обоих показателей).
Применение эволокумаба по сравнению с плацебо приводило к статистически значимому снижению основного комбинированного показателя смертности от осложнений ССЗ, частоты развития ИМ, инсульта, госпитализаций по поводу нестабильной стенокардии или выполнения реваскуляризации миокарда. Неблагоприятные исходы, включенные в основной показатель, в группе эволокумаба и группе плацебо развились у 9,8 и 11,3% больных соответственно (отношение риска 0,85 при 95% ДИ от 0,79 до 0,92; p<0,001). Кроме того, в группе эволокумаба по сравнению с группой плацебо отмечалось сходное снижение главного дополнительного комбинированного показателя смертности от осложнений ССЗ, частоты развития ИМ или инсульта. Неблагоприятные клинические исходы, включенные в такой показатель, в группе эволокумаба и группе плацебо развились у 5,9 и 7,4% больных соответственно (отношение риска 0,80 при 95% ДИ от 0,73 до 0,88; p<0,001). Отмечена тенденция к увеличению выраженности снижения основного показателя со временем с 12% (при 95% ДИ от 3 до 20%) в течение первого года наблюдения до 19% (при 95% ДИ от 11 до 27%) в последующем. Кроме того, отмечено сходное увеличение со временем выраженности снижения риска развития неблагоприятных исходов, включенных в главный дополнительный показатель, с 16% (при 95% ДИ от 4 до 26%) в течение первого года до 25% (при 95% ДИ от 15 до 34%) в последующем.
Результаты анализа отдельных компонентов оцениваемых показателей свидетельствовали об отсутствии влияния применения эволокумаба на смертность от осложнений ССЗ, а следовательно, и для других исходов при оценке значения p следует учитывать поисковый характер анализа отдельных компонентов комбинированных показателей. Риск развития ИМ, инсульта и выполнения реваскуляризации снижался на 21—27% в отсутствие влияния на частоту госпитализаций по поводу нестабильной стенокардии, смертность от осложнений ССЗ или частоту госпитализаций по поводу утяжеления СН, а также на общую смертность.
Преимущества применения эволокумаба по сравнению с плацебо по влиянию на основной и главный дополнительный показатели в основном были сходными в подгруппах больных в зависимости от определенных характеристик, включая возраст, пол и тип сосудистого заболевания, обусловленного атеросклерозом. Преимущества также были сопоставимыми у больных с исходными уровнями ХС ЛПНП, соответствующими разным квартилям, включая больных, у которых исходная концентрация ХС ЛПНП соответствовала верхнему квартилю (3,3 ммоль/л; МКД от 3 до 3,7 ммоль/л), и больных, у которых исходный уровень ХС ЛПНП соответствовал нижнему квартилю (1,9 ммоль/л; МКД от 1,8 до 2 ммоль/л). Преимущества применения эволокумаба также были устойчивыми при разной интенсивности терапии статинами, а также независимо от применения эзетимиба и при использовании обоих режимов дозирования эволокумаба (140 мг каждые 2 нед или 420 мг 1 раз в месяц).
Не было отмечено статистически значимых различий между группами в целом по частоте развития НЯ, включая тяжелые НЯ или НЯ, которые могли считаться обусловленными применением исследуемого препарата и приводили к прекращению его использования. Аналогично частота развития осложнений, обусловленных токсическим влиянием на мышцы, а также развития катаракты, нейрокогнитивных НЯ и геморрагического инсульта статистически значимо не различалась между группами. Реакции в месте инъекции исследуемого препарата отмечались редко, но в группе эволокумаба чаще, чем в группе плацебо (у 2,1 и 1,6% больных соответственно). Большинство таких реакций (примерно 90% в каждой группе) считались слабовыраженными и только у 0,1% больных в каждой группе прекратили применение исследуемого препарата в связи с развитием реакций в месте инъекции. Частота развития подтвержденных случаев развития СД статистически значимо не различалась между группами (отношение риска 1,05 при 95% ДИ от 0,94 до 1,17). Частота развития аллергических реакций также статистически значимо не различалась между группами (такие реакции в группе эволокумаба и группе плацебо отмечались у 3,1 и 2,9% больных соответственно). В группе эволокумаба антитела к эволокумабу определялись у 0,3% больных и ни в одном случае не выявлялись нейтрализующие антитела.
Репата (Repatha)
Механизм действия
Эволокумаб селективно связывается с пропротеиновой конвертазой субтилизин/кексин типа 9 (PCSK9) и препятствует связыванию циркулирующей PCSK9 с рецепторами липопротеинов низкой плотности (Р-ЛПНП) на поверхности гепатоцитов, тем самым предотвращая PCSK9- опосредованную деградацию Р-ЛПНП. Повышение содержания печеночных Р-ЛПНП приводит к соответствующему снижению концентрации холестерина липопротеинов низкой плотности (ХС-ЛПНП) в сыворотке крови.
Фармакодинамические эффекты
У пациентов с первичной гиперхолестеринемией и смешанной дислипидемией препарат Репата снижает концентрации несвязанной PCSK9, ХС-ЛПНП, общего холестерина (ОХС), аполипопротеина В (АпоВ), холестерина, не связанного с липопротеинами высокой плотности (ХС-не-ЛПВП), холестерина липопротеинов очень низкой плотности (ХС-ЛПОНП), триглицеридов (ТГ) и липопротеина(а) (Лп[а]), а также повышает концентрации холестерина липопротеинов высокой плотности (ХС-ЛПВП) и аполипопротеина А1 (AnoAl), улучшая соотношение ОХС/ХС-ЛПВП, АпоВ/АпоА1.
Однократное подкожное введение Репата в дозе 140 мг или 420 мг через 4 часа обеспечивает максимальное подавление циркулирующей несвязанной PCSK9 с последующим снижением ХС-ЛПНП, концентрация которого в среднем достигает минимума на 14 и 21 день соответственно. Изменения концентрации несвязанной PCSK9 и сывороточных липопротеинов обратимы после прекращения терапии Репата. Не отмечено повышения концентраций несвязанной PCSK9 или ХС-ЛПНП выше исходной концентрации в период вымывания эволокумаба. Это позволяет предположить, что во время лечения компенсаторные механизмы по увеличению выработки PCSK9 и ХС-ЛПНП не активируются.
Схемы терапии Репата с подкожным введением 140 мг 1 раз в 2 недели и 420 мг 1 раз в месяц были эквивалентны по среднему снижению ХС-ЛПНП (среднее значение через 10-12 недель), концентрация которого снижалась на 57-72% от исходного значения по сравнению с плацебо. Лечение Репата привело к аналогичному снижению ХС-ЛПНП при применении в виде монотерапии или в комбинации с другими гиполипидемическими средствами.
Клиническая эффективность и безопасность
Клиническая эффективность при первичной гиперхолестеринемии и смешанной дислипидемии
В результате терапии эволокумабом на неделе 1 было достигнуто снижение концентрации ХС-ЛПНП приблизительно на 55-75% и сохранялось на протяжении всего периода проведения долгосрочной терапии. Максимальный ответ, как правило, достигается через 1-2 недели при использовании режима 140 мг 1 раз в 2 недели и 420 мг 1 раз в месяц.
Препарат Репата был эффективен во всех подгруппах по сравнению с плацебо и эзетимибом, при отсутствии значимых различий между отдельными подгруппами, выделенными в зависимости от возраста, расы, пола, географического региона, индекса массы тела, категории риска по данным Национальной образовательной программы по холестерину, текущего статуса курения, исходных факторов риска ишемической болезни сердца (ИБС), наличия ранней ИБС в семейном анамнезе, статуса толерантности к глюкозе (т. е., сахарный диабет 2 типа, метаболический синдром или отсутствие обоих состояний), артериальная гипертензия, артериальной гипертензии, дозы и интенсивности терапии статинами, исходной концентрации несвязанной PCSK9, ХС-ЛПНП и триглицеридов.
У 80-85% всех пациентов с первичной гиперлипидемией, получавших эволокумаб в любой дозировке, наблюдалось снижение концентрации ХС-ЛПНП на ≥ 50% в среднем через 10-12 недель терапии. До 99% пациентов, которые получали Репата в любой дозе, достигали концентрации ХС-ЛПНП < 2,6 ммоль/л, и до 95% достигали концентрации ХС-ЛПНП <1,8 ммоль/л в среднем через 10-12 недель терапии.
Применение в комбинации со статином или со статином и другими гиполипидемическими средствами
Исследование LAPLACE-2 являлось исследованием длительностью 12 недель, в которое было включено 1896 пациентов с первичной гиперхолестеринемией или смешанной дислипидемией.
Препарат Репата обеспечивал достоверное снижение концентрации ХС-ЛПНП до средних значений через 10-12 недель относительно исходного уровня по сравнению с плацебо в группах розувастатина и симвастатина и по сравнению с плацебо и эзетимибом в группе аторвастатина (р < 0,001). Репата обеспечивал достоверное снижение ОХС, АпоВ, ХС-не-ЛПВП, ОХС/ХС-ЛПВП, АпоВ/АпоА1, ХС-ЛПОНП, ТГ и Лп(а), а также повышение концентрации ХС-ЛПВП (среднее значение через 10-12 недель относительно исходного уровня) по сравнению с плацебо в группах розувастатина и симвастатина (р < 0,05) и достоверно снижал концентрации ОХС, АпоВ, ХС-не-ЛПВП, ОХС/ХС- ЛПВП, АпоВ/АпоА1 и Лп(а) по сравнению с плацебо и эзетимибом в группе аторвастатина (р < 0,001).
Исследование RUTHERFORD-2 являлось исследованием длительностью 12 недель, в которое было включено 329 пациентов с гетерозиготной семейной гиперхолестеринемией, получавших гиполипидемическую терапию.
Препарат Репата достоверно снижал концентрацию ХС-ЛПНП до средних значений через 10-12 недель относительно исходного уровня по сравнению с плацебо (р < 0,001). Репата значимо снижал ОХС, АпоВ, ХС- не-ЛПВП, ОХС/ХС-ЛПВП, АпоВ/АпоА1, ХС-ЛПОНП, ТГ и Лп(а), а также повышал концентрации ХС-ЛПВП и AnoAl до средних значений на 10-12 неделе относительно исходного уровня по сравнению с плацебо (р < 0,05).
Пациенты с непереносимостью статинов
Исследование GAUSS-2 являлось исследованием длительностью 12 недель, в которое было включено 307 пациентов с полной непереносимостью статинов или непереносимостью эффективной дозы статинов. Репата значимо снижал ХС-ЛПНП по сравнению с эзетимибом (р < 0,001). Репата значимо снижал ОХС, АпоВ, ХС-не-ЛПВП, ОХС/ХС-ЛПВП, АпоВ/АпоА1 и Лп(а) до средних значений на 10-12 неделе относительно исходного уровня по сравнению с эзетимибом (р < 0,001).
Лечение в отсутствии статинов
Исследование MENDEL-2 являлось исследованием препарата Репата длительностью 12 недель, в котором приняли участие 614 пациентов с первичной гиперхолестеринемией и смешанной дислипидемией. Репата значимо снижал ХС-ЛПНП до средних значений через 10-12 недель относительно исходного уровня по сравнению с плацебо и эзетимибом (р < 0,001). Репата значимо снижал ОХС, АпоВ, ХС-не-ЛПВП, ОХС/ХС-ЛПВП, АпоВ/АпоА1 и Лп(а) до средних значений через 10-12 недель относительно исходного уровня по сравнению с плацебо и эзетимибом (р < 0,001).
Эффективность длительной терапии при первичной гиперхолестеринемии и смешанной дислипидемии
Исследование DESCARTES являлось исследованием длительностью 52 недели, в которое был включен 901 пациент с гиперлипидемией, получавший только диету, аторвастатин или комбинацию аторвастатина и эзетимиба. Применение препарата Репата в дозе 420 мг 1 раз в месяц обеспечивало достоверное снижение концентрации ХС-ЛПНП через 52 недели относительно исходного уровня по сравнению с плацебо (р< 0,001). Эффекты лечения сохранялись в течение 1 года, о чем свидетельствует снижение концентрации ХС-ЛПНП с 12 до 52 недели. На фоне гиполипидемической терапии, оптимизированной по концентрации ХС-ЛПНП и сердечно-сосудистому риску, отмечалось постоянное снижение концентрации ХС-ЛПНП в течение 52 недель относительно исходного уровня по сравнению с плацебо.
Репата значимо снижал ОХС, АпоВ, ХС-не-ЛПВП, ОХС/ХС-ЛПВП, АпоВ/АпоА1, ХС-ЛПОНП, ТГ и Лп(а), а также повышал концентрации ХС-ЛПВП и AnoAl на 52 неделе по сравнению с плацебо (р < 0,001). OSLER и OSLER-2 — два исследования по долгосрочной оценке безопасности и эффективности Репаты у пациентов, завершивших лечение в основном исследовании. Всего в исследование OSLER было включено 1324 пациента. Репата в дозе 420 мг 1 раз в месяц значимо снижал ХС-ЛПНП через 12 недель и через 52 недели относительно исходного уровня по сравнению с контролем (номинальное р < 0,001). Эффекты лечения сохранялись в течение 272 недель, о чем свидетельствует снижение концентрации ХС-ЛПНП с 12-й недели в основном исследовании до 260-й недели в открытом дополнительном исследовании. Всего в исследование OSLER-2 был включен 3681 пациент. Применение препарата Репата обеспечивало достоверное снижение концентрации ХС-ЛПНП через 12 недель и через 48 недель относительно исходного уровня по сравнению с контролем (номинальное р< 0,001). Эффекты лечения сохранялись, о чем свидетельствует снижение концентрации ХС-ЛПНП с 12-й недели до 104-й недели в открытом дополнительном исследовании.
Применение препарата Репата обеспечивало достоверное снижение концентраций ОХС, АпоВ, ХС-не-ЛПВП, ОХС/ХС-ЛПВП, АпоВ/АпоА1, ХС-ЛПОНП, ТГ и Лп(а), а также увеличивал ХС-ЛПВП и AnoAl относительно исходного уровня на 52-й неделе в исследовании OSLER и на 48-й неделе в исследовании OSLER-2 по сравнению с контролем (номинальное р < 0,001). ХС-ЛПНП и другие параметры липидного обмена вернулись к исходным значениям в течение 12 недель после отмены Репата в начале исследования OSLER или OSLER-2 без признаков рикошета.
TAUSSIG — исследование для оценки долгосрочной безопасности и эффективности препарата Репата, применяемого в качестве дополнения к другой гиполипидемической терапии у пациентов с тяжелой семейной гиперхолестеринемией (СГХС), включая гомозиготную семейную гиперхолестеринемию.
TAUSSIG — продленное исследование длительностью 5 лет для оценки долгосрочной безопасности и эффективности препарата Репата, применяемого в качестве дополнения к другой гиполипидемической терапии у пациентов с тяжелой семейной гиперхолестеринемией (СГХС), включая гомозиготную семейную гиперхолестеринемию. Всего в исследование TAUSSIG были включены 194 пациента с тяжелой семейной гиперхолестеринемией (не-ГоСГХС) и 106 пациентов с гомозиготной семейной гиперхолестеринемией. Все пациенты в исследовании первоначально получали препарат Репата в дозе 420 мг 1 раз в месяц, за исключением тех, кто получал аферез липидов на момент включения в исследование; эти пациенты начинали получать препарат Репата с дозы 420 мг 1 раз в 2 недели. Частота дозирования у пациентов, не получающих аферез, могла повышаться до 420 мг 1 раз в 2 недели в зависимости от ответа концентраций ХС-ЛПНП и PCSK9. Длительное применение препарата Репата обеспечило стойкий терапевтический эффект, о чем свидетельствовало снижение концентрации ХС-ЛПНП у пациентов с тяжелой семейной гиперхолестеринемией (не-ГоСГХС). Изменение других параметров липидного обмена (ОХС, АпоВ, ХС-не-ЛПВП, ОХС/ХС-ЛПВП и АпоВ/АпоА1) также указывало на стойкий эффект длительного применения Репата у пациентов с тяжелой семейной гиперхолестеринемией (не-ГоСГХС).
Долгосрочная безопасность поддержания ХС-ЛПНП на очень низком уровне (т. е. < 0,65 ммоль/л [< 25 мг/дл]) еще не установлена. Имеющиеся данные показывают, что клинически значимые различия между профилем безопасности пациентов с концентрацией ХС-ЛПНП <0,65 ммоль/л и пациентов с более высокой концентрацией ХС-ЛПНП не отмечаются (см. раздел «Побочное действие»).
Лечение гомозиготной семейной гиперхолестеринемии
Исследование TESLA являлось исследованием длительностью 12 недель с участием 49 пациентов, страдавших гомозиготной семейной гиперхолестеринемией в возрасте от 12 до 65 лет.
Применение препарата Репата в дозе 420 мг 1 раз в месяц в качестве дополнения к другим видам гиполипидемической терапии (например, статинам, секвестрантам желчных кислот) значимо снижало ХС-ЛПНП и АпоВ через 12 недель по сравнению с плацебо (р< 0,001). Изменения других параметров липидного обмена (ОХС, ХС-не-ЛПВП, ОХС/ХС-ЛПВП и АпоВ/АпоА1) также указывали на терапевтический эффект применения препарата Репата у пациентов с гомозиготной семейной гиперхолестеринемией.
Эффективность длительной терапии при гомозиготной семейной гиперхолестеринемии
В исследовании TAUSSIG длительное применение препарата Репата обеспечивало стойкий терапевтический эффект, о чем свидетельствует снижение концентрации ХС-ЛПНП приблизительно на 20-30% у пациентов с гомозиготной семейной гиперхолестеринемией, которые не проходили аферез, и приблизительно на 10-30% у пациентов с гомозиготной семейной гиперхолестеринемией на фоне афереза. Изменения других параметров липидного обмена (ОХС, АпоВ, ХС-не-ЛПВП, ОХС/ХС-ЛПВП и АпоВ/АпоА1) также указывали на стойкий эффект длительного применения Репата у пациентов с гомозиготной семейной гиперхолестеринемией. Снижение концентрации ХС-ЛПНП и изменения других параметров липидного обмена у 14 подростков (в возрасте от ≥ 12 до < 18 лет) с гомозиготной семейной гиперхолестеринемией сопоставимы с таковыми в общей популяции пациентов с данной патологией.
Эффективность при атеросклеротических заболеваниях
Влияние препарата Репата (в дозе 420 мг 1 раз в месяц) у пациентов с атеросклеротическими заболеваниями, измеряемое с помощью внутрисосудистого ультразвукового исследования (ВСУЗИ), оценивалось в рамках 78-недельного исследования с участием 968 пациентов с ишемической болезнью сердца на фоне стабильной оптимальной терапии статинами. Препарат Репата уменьшал как относительный объем атеромы (PAV; 1,01% [95% ДИ 0,64-1,38], р< 0,0001), так и общий объем атеромы (TAV; 4,89 мм3 [95% ДИ 2,53-7,25], р < 0,0001) по сравнению с плацебо. Регрессия атеросклеротических поражений согласно данным PAV наблюдалась у 64,3% (95% ДИ 59,6-68,7) и 47,3 % (95% ДИ 42,6-52,0) пациентов, получавших Репата или плацебо соответственно. При измерении TAV регрессия атеросклеротических поражений наблюдалась у 61,5% (95 % ДИ 56,7-66,0) и 48,9% (95% ДИ 44,2-53,7) пациентов, получавших Репата или плацебо соответственно. В исследовании не изучалась корреляция между регрессией атеросклеротического заболевания и частотой сердечно-сосудистых событий.
Снижение риска сердечно-сосудистых событий у взрослых с диагностированными сердечно-сосудистыми заболеваниями, обусловленными атеросклерозом
Исследование результатов терапии препаратов Репата (FOURIER) представляло собой рандомизированное двойное слепое исследование, основанное на оценке целевых событий, в котором приняли участие 27 564 пациента в возрасте от 40 до 86 лет (средний возраст 62,5 года) с установленным атеросклеротическим сердечно-сосудистым заболеванием; 81% имели предшествующий инфаркт миокарда (ИМ), 19% — инсульт и 13% — заболевания периферических артерий. Более 99% пациентов получали терапию статинами средней и высокой интенсивности и по крайней мере еще одно сердечно-сосудистое лекарственное средство, например, антитромбоцитарные препараты, бета-блокаторы, ингибиторы АПФ или блокаторы рецепторов ангиотензина; медианная (Ql, Q3) исходная концентрация ХС-ЛПНП составляла 2,4 ммоль/л (2,1, 2,8). Абсолютный сердечно-сосудистый риск был сбалансирован между группами лечения: в дополнение к индексному событию у всех пациентов отмечались по крайней мере 1 основной или 2 малозначительных фактора сердечно-сосудистого риска; 80% имели артериальную гипертензию, 36% сахарный диабет и 28% ежедневно курили. Пациенты были рандомизированы в соотношении 1:1 и получали либо Репата (140 мг 1 раз в 2 недели или 420 мг 1 раз в месяц), либо соответствующее плацебо; средняя продолжительность наблюдения за пациентом составила 26 месяцев.
На протяжении всего исследования наблюдалось значимое снижение концентрации ХС-ЛПНП, при этом медиана диапазона ХС-ЛПНП достигала 0,8-0,9 ммоль/л при каждой оценке; 25 % пациентов достигли концентрации ХС-ЛПНП менее 0,5 ммоль/л. Несмотря на достигнутый очень низкий уровень ХС-ЛПНП, новых проблем с безопасностью не наблюдалось (см. раздел «Побочное действие»); частота новых случаев сахарного диабета и когнитивных нарушений была сопоставимой у пациентов с ХС-ЛПНП < 0,65 ммоль/л и с более высокой концентрацией ХС-ЛПНП.