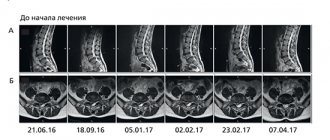
Detailed description of the study
Alpha1-antitrypsin (AAT) is a protein whose function is to protect the lungs and prevent the destruction of their alveoli. The formation of AAT is carried out by hepatocytes, or liver cells. Entering the blood, this protein penetrates the alveoli of the lungs. The main biochemical role of the AAT molecule is to suppress the activity of neutrophil enzymes, for example, trypsin, elastase, proteinase, which are released during normal phagocytosis - the capture and inactivation of foreign particles in the lungs.
AAT deficiency is a relatively common inherited disorder. The production of this protein is controlled by genes in the protease inhibitor locus. The SERPINA1 gene, responsible for encoding AAT, is located on chromosome 14 and is associated with more than 100 variants. M alleles are the most common and normal variants. Most patients with clinical manifestations of AAT deficiency are homozygous SS or ZZ or heterozygous MS, MZ or SZ.
In persons with a genetic defect of AAT, there is a decrease in the synthesis of this protein in the liver, and as a result, its low concentration in the blood serum and alveoli. An imbalance between proteases and antiproteases in the alveoli leads to unimpeded destruction of elastin and collagen proteins in the walls of the alveoli by neutrophils. In addition, the accumulation of altered proteins in the liver instead of AAT leads to damage and destruction of hepatocytes.
Damage to the lungs and liver may vary in severity and time of clinical manifestations depending on the degree of AAT deficiency. With some SERPINA1 gene mutations, symptoms begin as early as infancy. Mild AAT deficiency may not be clinically significant if the mutation carrier does not have other factors causing lung tissue damage.
The most common factors contributing to the progression of changes in the lungs due to AAT deficiency is smoking. The entry of tobacco smoke and tar into the respiratory system leads to the activation of a large number of neutrophils and accelerated destruction of the alveoli. On average, cigarette smoking shortens the onset of the disease by about 10 years.
Other factors that can hasten the onset or worsen symptoms include exposure to dust or hot steam, which is often occupational.
The lung disease resulting from AAT deficiency is emphysema. It is characterized by pathological expansion of the smallest bronchi and alveoli with their damage and destruction. The consequence of these morphological changes is a decrease in the respiratory surface of the lungs and a decrease in blood oxygen saturation. Slowly progressive shortness of breath is the primary symptom, although some patients first present with a cough producing sputum. Signs of lung damage may appear at a young age, which determines the need to assess AAT levels in patients with emphysema.
Along with changes in the respiratory system, damage to liver tissue is observed. It may initially manifest as laboratory abnormalities but then, if left untreated, progress to chronic hepatitis, followed by fibrosis and cirrhosis. Inflammation of the liver tissue sometimes does not manifest itself until the final stage of changes in this organ—cirrhosis—develops. A person may notice a deterioration in general well-being, indigestion, increased gas formation, an increase in abdominal volume along with a decrease in muscle mass in the extremities, a tendency to bruise, redness of the palms, hair loss and many other symptoms.
In most cases, liver damage is observed in the second half of life and can often be regarded as fatty degeneration or other more common diseases of this organ. Assessment of AAT levels is a mandatory step in the diagnosis of liver diseases, especially if no obvious cause of the disease has been identified.
Alpha-1 antitrypsin deficiency
Alpha-1 antitrypsin deficiency. What is this?
Alpha-1 antitrypsin deficiency is a rare genetic disorder that occurs primarily in childhood. The first symptoms may appear several months after birth; subsequently, the clinical picture changes, and the disease takes on two forms. The most common is pulmonary, it affects 88% of patients with alpha-1 antitrypsin deficiency. The other form is the hepatic form, which affects the remaining 12% of patients with alpha-1 antitrypsin deficiency.
Doctors may write the disease as follows: α1-antitrypsin deficiency, α1-antitrypsin deficiency, alpha-1-antitrypsin deficiency. As its name suggests, the disease is caused by a deficiency in the production of the α1-antitrypsin protein, and all its clinical manifestations are associated with a deficiency of this protein.
Alpha-1 antitrypsin deficiency. Causes of the disease.
Alpha-1-antitrypsin - belongs to the proteins of the acute phase of inflammation, produced in the liver. Alpha-1 protease inhibitor (α-1-AT) is related to albumin, its molecular weight is 50,000-55,000. This protein causes 90-92% of the total antiprotease activity of blood plasma and has a fairly wide range of physiological and pharmacological effects. Thus, it inhibits the activity of enzymes such as chymotrypsin, trypsin, plasmin, thrombin, elastase, kallikrein and collagenase. Its main physiological function is the inhibition of neutrophil protease, elastase, which hydrolyze structural proteins.
Simply put, it is a protein that is produced by liver cells. Its main function is the breakdown of proteases (they break down food particles into molecules, fight microbes, and protect against adverse environmental factors). And if, due to a “broken” gene, we are faced with a deficiency of alpha-1 antitrypsin, then there are too many proteases, and they begin to destroy the lungs. In this case, alpha-1 antitrypsin does not enter the blood, but remains in the liver, which also provokes its destruction.
As a result, with a rare genetic disease, alpha-1 antitrypsin deficiency, two pathological processes are formed:
- Pulmonary. The interalveolar septa that form the lung tissue are destroyed. Because of this, gas exchange begins to suffer, and, with the development of the pathological process, pulmonary emphysema may develop after 30 years.
- Hepatic. In the liver, in which excess alpha-1 antitrypsin remains, scar tissue begins to form, which in adulthood leads to chronic liver failure, cirrhosis.
The course of the disease can be influenced not only by genetic, but also by external factors. Thus, living in large cities, with increased air pollution and gases, leads to the acceleration of pathological processes. Constant interaction with construction dust and work in hazardous chemical industries are also considered dangerous. There is a clear connection between emphysema and smoking. Clinical practice shows that in a smoker with alpha-1 antitrypsin deficiency, the lungs begin to fail approximately 10 years earlier than in a non-smoker.
Alpha-1 antitrypsin deficiency. Inheritance.
The disease is inherited in an autosomal codominant manner, which means it is definitely transmitted to children. The severity of alpha-1 antitrypsin deficiency depends on whether the child inherited one or two broken genes. Thus, the most severe course of the disease is observed in children where both parents have alpha-1 antitrypsin deficiency.
Alpha-1 antitrypsin deficiency. Symptoms of the disease.
Symptoms of the disease that appear in childhood:
- cough;
- wheezing;
- slowly progressive shortness of breath;
- prolonged jaundice;
- retardation in physical development.
Symptoms of the disease that appear in adulthood (after 30-40 years):
- cough, which produces copious amounts of dark mucus;
- a person's breathing becomes wheezing;
- the alternation of inhalation and exhalation is disrupted;
- a person is susceptible to frequent inflammatory diseases that affect both the upper and lower respiratory tract;
- scarring of liver tissue;
- cirrhosis.
Alpha-1 antitrypsin deficiency. Diagnostics.
Like most rare genetic diseases, alpha-1 antitrypsin deficiency is very difficult to detect at an early stage; sometimes it takes years to understand that impaired liver or lung function is a manifestation of a “broken” gene.
The diagnosis is made on the basis of medical history, the presence in the family of patients with pulmonary pathology or non-infectious liver damage. Typical complaints must be considered and the lack of effect of the therapy taken into account.
Laboratory diagnosis of the disease occurs by determining the level of alpha-1 antitrypsin deficiency in the blood. The concentration of α-1-antitrypsin in the blood serum depends significantly on the age of the patient; normally it usually ranges from 110-200 mg/dl (SI: 1.1-2 g/l). When it decreases to 0.8 g/l (11 mmol/l), the presence of the disease can be indicated.
If alpha-1 antitrypsin deficiency is detected in the laboratory, genetic testing must be performed. It reveals characteristic mutations (changes in the structure of chromosomes), which, depending on the degree of alpha-1 antitrypsin deficiency, are coded as: PiMM: 100% (normal), PIMS (80% of the normal level), PiSS (60% of the normal level) , PiMZ (60%), PiSZ (40%), PiZZ (10-15%).
All other diagnostic methods are aimed at determining the degree of damage to the tissues of the lungs and liver. These are various types of tests, the result of which shows lung volume and respiratory frequency; X-ray examination of the condition of the tissues of the lungs and liver; tomography to determine the extent of organ damage; blood tests and spirometric readings are taken.
Acquired forms of alpha-1 antitrypsin deficiency are possible in nephrotic syndrome, gastroenteropathy with protein loss, and thermal burns in the acute phase. A decrease in the content of alpha-antitrypsin in the blood serum can be diagnosed in patients with viral hepatitis as a violation of its formation in the liver. Increased consumption of this glycoprotein is observed in respiratory distress syndromes, coagulopathies, acute pancreatitis, which also causes a decrease in its concentration in the blood serum. All patients with chronic liver pathology are recommended to routinely determine the level of alpha-antitrypsin; this is due to the impossibility of establishing a correct and final diagnosis only on the basis of clinical data alone. Corticosteroids, oral contraceptives, and eating or smoking before the test lead to a false increase in alpha-antitrypsin levels.
Alpha-1 antitrypsin deficiency. Treatment.
Rare genetic diseases cannot be treated; it is only possible to use different methods of influence to limit the manifestations of the destructive factors of the disease.
To date, the only available specific treatment for patients with the pulmonary form of A1AT deficiency is intravenous replacement (augmentation) therapy with human A1AT obtained from a pool of donor plasma.
Replacement therapy slows the progression of lung disease and is the most pathogenetically substantiated method of treating A1AT deficiency.
Replacement therapy effectively reduces the annual loss of lung tissue, as evidenced by the assessment of CT densitometry indicators over time. There is also evidence of a decrease in the rate of decline in FEV1 per year and a statistically significant increase in life expectancy with augmentation therapy. According to a number of studies, replacement therapy helps reduce the frequency and severity of exacerbations of COPD with A1AT deficiency, as well as improve the quality of life of patients
Replacement therapy is aimed at achieving and maintaining A1AT concentrations above the safety threshold of 0.8 g/l. Correcting the deficiency cannot restore damaged lung structures, but it can prevent further damage to lung tissue and stabilize the patient's condition.
In addition, the following are indicated: antibacterial drugs, inhaled bronchodilators, cephalosporins, mucolytics, antifungals, antipyretics and hepatoprotective agents.
In severe and advanced cases, surgical interventions are necessary. Organ transplantation may be indicated.
Alpha-1 antitrypsin deficiency. Prevention.
To prevent the progression of the disease, it is recommended to avoid adverse effects on the lungs and liver. To do this you need:
- Stop smoking. Do not go near places where other people smoke. Try not to inhale tobacco smoke even passively;
- Ventilate the premises frequently to ensure a constant flow of fresh air;
- Do not contact people with infectious diseases. Carry out preventive measures to avoid getting sick yourself;
- Do not go outside in windy weather. Do not walk along busy highways and roads, in places where there is increased air pollution;
- Avoid alcohol as a substance that has an extremely harmful effect on the liver;
- Complete all preventive vaccinations. During epidemics, wear a mask and wash your hands frequently;
- Try to choose a place to live with a good environmental situation: a small town without industrial enterprises or a village;
- Follow a gentle diet, do not eat smoked, spicy, fatty, or fried foods. Eat more seafood;
- Do not carry out construction work yourself that involves a large amount of dust;
- Limit serious physical activity. Physical therapy is acceptable. Special breathing exercises are desirable;
- Take hepatoprotectors periodically.
Alpha-1 antitrypsin deficiency is an incurable disease, so medical supervision must be carried out for life. It is very important to take medications in a timely manner and follow all recommendations to minimize the pathological impact of the disease.
Useful addresses:
— genetic diagnosis of alpha-1 antitrypsin deficiency can be done at the Federal State Budgetary Institution “Medical Genetic Research Center”, Moscow, st. Moskvorechye, 1
— treatment of patients with alpha-1 antitrypsin deficiency is carried out at the Research Institute of Pulmonology of the Federal Medical and Biological Agency of Russia, Moscow, st. 11th Parkovaya, 32/61
If you have any questions, you can always contact us by calling our hotline (499) 270 3520