Introduction
In the field of practical pediatric hepatology, a special place is occupied by orphan diseases that occur with liver damage, which, in particular, include glycogen storage disease (GBD). In our country, the experience of supervising such patients due to the rarity of this nosology is relatively small, and accordingly, there is not a sufficient number of specialist doctors in this field. The clinical and laboratory picture of the disease is extremely diverse, which often complicates its timely diagnosis and leads to the development of severe complications and disability of patients. In this regard, it seems relevant to present in this article modern information on the etiology, pathogenesis, clinical picture, methods of diagnosis and treatment of hypertension in children.
Classification
The structure of the concept of “glycogen disease” includes at least 15 types of genetically determined pathological conditions, which are based on a violation of the synthesis or breakdown of glycogen with the accumulation of the latter in various organs and tissues, mainly in the liver and muscles. Currently, it is customary to use the classification proposed by G. Cori in 1954 and built on a chronological principle: types of HD are designated by Roman numerals and are arranged in the order of discovery of the phenotype and the corresponding enzyme defects [1]. However, the classification is constantly revised, which causes difficulties in making differential diagnoses between different types of the disease. For example, there is currently no division into subtypes VIa and VIb. Type VIII GB is also excluded from the nomenclature. At the same time, the classification of type IX has been significantly expanded, in the structure of which 5 subtypes are identified (a1, a2, b, c, d). In addition, new types have been added to the classification - XIV, XV. Previously, index X was assigned to a disease associated with deficiency of cyclic 3,5-AMP-dependent kinase (described in a single patient), but subsequently this form of pathology was classified as type VI. Currently, subtype X is a disease caused by deficiency of muscle phosphoglycerate mutase-2. Although glycogen synthase deficiency does not result in excessive accumulation of glycogen in the liver, but rather in extremely low levels of glycogen, this form of pathology is often classified as HD type 0 because it is also a defect in glycogen metabolism and causes changes similar to other forms of HD. It is important to note that one patient may have a combination of several enzyme defects, and in such cases it is customary to talk about unidentified types of this disease [2]. In this article we will touch only on those types of headache that occur with predominant liver damage (types I, III, VI and IX).
Etiology
Glycogen disease type Ia is caused by mutations in the G6PC gene encoding glucose-6-phosphatase, which leads to its deficiency in the liver, kidneys, intestinal mucosa [3, 4], and also, according to some data, in the islets of pancreatic β-cells glands and gallbladder [5]. Type Ib headache is caused by mutations in the SLC17A4 gene, which encodes microsomal transport protein T1 (glucose-6-phosphatase translocase), which leads to its deficiency in the liver, kidneys, and intestinal mucosa [6]. Glycogen disease type III occurs as a result of mutations in the AGL gene, which encodes a glycogen debranching enzyme, which has two catalytic units: amylo-1,6-glucosidase and 4-alpha-glucanotransferase, capable of functioning independently of each other, but for the normal action of the debranching enzyme activity of both catalytic units is required. The vast majority of patients have a deficiency of this enzyme in both the liver and muscle (subtype IIIa), but in approximately 15% of patients it is observed only in the liver (subtype IIIb). The presence of these subtypes is explained by different expression of the enzyme in tissues [7–10].
Type VI headache is caused by mutations in the PYGL gene, which encodes liver phosphorylase [11, 12]. Glycogen storage disease type IX develops against the background of mutations in the PHKA2 gene - for IXa1, IXa2 subtypes, in PHKAB - for the IXb subtype [13, 14].
Pathogenesis
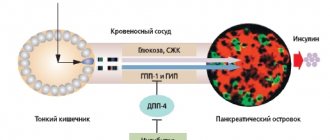
A description of the main links in the pathogenesis and clinical and laboratory manifestations of type I hypertension can serve as the basis for understanding the symptoms of all other hepatic forms of this pathology. The cause of the development of type I headache is a hereditary defect in glucose-6-phosphatase, an enzyme that ensures the release of glucose into the bloodstream after its release from liver cell glycogen. Thus, the reaction of the formation of free glucose from glucose-6-phosphate is disrupted, resulting in hypoglycemia. In addition, due to a defect in glucose-6-phosphatase, the substrate of the blocked reaction, glucose-6-phosphate, accumulates in liver cells, which, being involved in the catabolism process, is converted into pyruvate and lactate. At the same time, the level of lactate in the blood increases significantly, so the development of metabolic lactic acidosis is possible. In severe cases, hypoglycemia can result in seizures, coma, and death. Hypoglycemia is accompanied by a decrease in insulin levels and a decrease in the insulin/glucagon ratio, which in turn leads to accelerated lipolysis of adipose tissue (as a result of the action of glucagon) and the release of fatty acids into the blood. The concentration of triglycerides in the blood increases as a result of a decrease in the activity of lipoprotein lipase of adipose tissue, an enzyme activated by insulin and ensuring the absorption of triglycerides by adipose tissue cells. At the same time, against the background of a significantly increasing content of glucose-6-phosphate in cells, its use in the pentose phosphate pathway increases with increased formation of ribose-5-phosphate, a substrate for the synthesis of purine nucleotides, the final product of which is uric acid. The excretion of uric acid with the formation of urates also decreases due to an increase in lactate production and a change in urine pH to the acidic side, which complicates the excretion of urates - sparingly soluble salts of uric acid [2, 15–18].
Clinical picture, laboratory manifestations
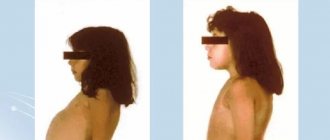
The onset of type I headache occurs in the first days after the birth of a child. The main symptom of the disease is severe hypoglycemia, which occurs even with the slightest fasting. Its clinical manifestations may be unmotivated prolonged crying and restlessness of the child, increased sweating, pallor of the skin, cyanosis of the nasolabial triangle, lethargy; convulsions may develop. Severe hepatomegaly soon follows, leading to a significant increase in the size of the abdomen, which may initially be mistaken for bloating or ascites and leads to an incorrect interpretation of the pathology. In children with type I hypertension, local deposits of subcutaneous fatty tissue may be observed, mainly on the cheeks (“doll” face), chest, buttocks, and thighs [3, 18]. With age, the disease progresses and multiple complications develop. Against the background of excessive glycogen deposition in the liver and pronounced dystrophic changes in hepatocytes, fibrosis is formed with the possibility of subsequent cirrhotic transformation. Liver adenomas can also occur, which have a potential risk of malignancy [19–21]. Chronic pancreatitis often develops against the background of hyperlipidemia [22, 23]. In the postpubertal period, hyperuricemia with its clinical complications in the form of gouty arthritis comes to the fore. Renal lesions in type I hypertension are manifested by focal segmental glomerulosclerosis, accompanied by proteinuria with decreased renal function, tubular disorders of the proximal type, reminiscent of DeToni-Debreu-Fanconi syndrome, nephrolithiasis and nephrocalcinosis [3, 5, 24]. Patients are stunted, have osteoporosis, and an increased tendency to fractures. Intermittent diarrhea, reminiscent of inflammatory bowel disease (Crohn's-like syndrome) may occur [25].
In children with type Ib hypertension, due to severe neutropenia (up to agranulocytosis), frequent recurrent infections with the development of purulent-septic foci are observed [26]. The prognosis for type I hypertension is relatively favorable only with early diagnosis and timely treatment. Without a special diet, frequent feedings and self-control, the prognosis is unfavorable - most patients die in early childhood from hypoglycemic coma, associated severe infections, and in older age - from hepatocellular carcinoma and gouty nephropathy [3, 18].
In type III hypertension, glycogen degeneration is impaired, which leads to the formation of pathological glycogen, which accumulates in hepatocytes and muscles and has a toxic effect. The clinical manifestations of type III hypertension are in many ways similar to those of type I: hepatomegaly (with cirrhosis, splenomegaly is added), growth retardation. In addition, cardiomegaly and muscle weakness are noted. Laboratory findings include hypoglycemia and hyperlipidemia, moderate acidosis and ketonuria, and cytolysis syndrome. The prognosis in most cases is favorable, but cases of disease progression with the development of liver cirrhosis and severe myopathy have been described [3, 18].
Type VI HD is characterized by hepatomegaly, hypoglycemia, hypercholesterolemia, and increased levels of transaminases in the blood serum. The prognosis is favorable, because disturbances in glycogen metabolism are compensated by gluconeogenesis. Manifestations of HD IX are almost identical to those of type VI [18].
Diagnostics
As already mentioned, hypertension can be suspected based on the totality of the described clinical and laboratory signs. However, final verification of the diagnosis requires a molecular genetic study, which requires specialized equipment and trained personnel, which entails large financial costs and does not yet allow this method to be widely used in clinical practice [3, 18]. At the same time, an important point is to assess the severity of liver fibrosis in children with hypertension. With routine ultrasound examination, it is impossible to distinguish between intermediate stages of hepatic fibrosis and it is not always possible to identify the initial signs of cirrhosis. The main method for diagnosing the severity of pathological changes in the liver is a puncture biopsy [27]. However, its use in pediatric practice is limited due to the invasiveness of the procedure and the need for anesthesia, which is not always possible for children with severe hypertension due to severe metabolic decompensation and a high risk of complications. In recent years, the role of serum markers of liver fibrosis has been actively discussed. The most studied are hyaluronic acid, type IV collagen, matrix metalloproteinases and their tissue inhibitors. They were shown to be informative in the diagnosis of various stages of liver fibrosis in children with chronic forms of its pathology, incl. and with hypertension [28, 29].
Treatment
Today, the most pressing problem for patients with hepatic forms of hypertension and their attending physicians is the lack of developed pathogenetic therapy, which inevitably affects the prognosis of the disease and the quality of life of patients. Therefore, the main method of treatment continues to be specific diet therapy, the main goal of which is to normalize carbohydrate metabolism and prevent secondary metabolic disorders by maintaining normoglycemia for as long as possible. In this regard, great importance is attached to the organization of fractional meals with an even distribution of easily soluble carbohydrates throughout the day; for this purpose, the number of meals is increased to 6–8 times a day (including early breakfast and late dinner). An integral component of the diet is the administration of raw corn starch, which has the property of being slowly and continuously broken down by pancreatic amylase into glucose, which prolongs the period of normoglycemia. Starch should be consumed every 4-6 hours (including nightly intake). Sucrose (table sugar), fructose and galactose are excluded from the diet, because These sugars in patients with hypertension are not converted to glucose, as in healthy people, but to lactate, which can aggravate lactic acidosis. Sugar-containing fruits (fresh and dried), all sweet confectionery products, juices, fruit waters, honey, and some medications are also strictly limited. Green apples, lemon, jam made with glucose, and vegetables such as cabbage, spinach, and leeks are allowed [30].
Ursodeoxycholic acid preparations (for example, Ursofalk, Ursosan) and essential phospholipids (for example, Essentiale-Forte N) can be used as hepatoprotective therapy. For secondary complications of hypertension, correction of tubular disorders, osteoporosis, and elimination of congestion in the gallbladder is carried out. The main therapeutic measure in the event of metabolic acidosis is the intravenous administration of alkaline solutions of sodium bicarbonate. For neutropenia, granulocyte colony-stimulating factor is used in children with type Ib hypertension [26].
A promising area of experimental medicine abroad is currently the study of the effectiveness of adenoviral vectors as a potential pathogenetic enzyme replacement therapy for hepatic forms of HD, which, however, is so far carried out only on animal models [31].
In recent years, data have emerged on successful liver transplantation in a small number of patients with HD [32]. In our country for 2009–2011.
The first such operations were performed on three children with type I of this pathology. Further observation of the patients showed positive results: laboratory parameters normalized, no cases of hypoglycemia were recorded, and lactic acidosis leveled out. Thus, the first successful clinical experience indicates the possibility of transplanting liver fragments from living related donors and opens up prospects for radical treatment of patients with type I glycogenosis in Russia [33, 34].
Currently, thanks to the development of effective diagnostic and treatment approaches, most chronic diseases can be effectively controlled and complications can be prevented. The full and timely use of these measures significantly prolongs the life of patients and also improves its quality. However, it is not possible to successfully control a chronic disease without the active participation of the patient himself. Mastering the skills to control and treat your disease requires special training with the participation of specialists, which is called patient education [35]. As with diabetes, self-control is extremely important with hypertension. Using modern methods of express analysis (glucometers, test strips), patients can independently assess the most important metabolic parameters with an accuracy close to laboratory ones. The goals of self-control are the prevention of complications, normal well-being, normal physical development and growth, a full life in society and a high quality of life. It should be remembered that the main meaning of self-control is not only regular checking of glycemic indicators, but also adequate assessment of the results, independent correction of treatment and planning of certain actions if the results of self-control are unsatisfactory [36, 37].
Currently, in our country, therapeutic training for patients with hypertension has not been developed, despite the fact that this is an extremely pressing problem in domestic healthcare. In this regard, it should be noted that any chronic disease reduces the quality of life of the child, as well as the family in which he is raised [38]. Therefore, the process of rehabilitation of children with hypertension should also include the assessment of this indicator. Recently, this concept has become an integral part of healthcare and has become firmly established in clinical and medical-social research. In foreign pediatrics, the quality of life indicator is included in the standards for examining patients, and is actively used in population studies to develop age-sex standards, carry out individual monitoring of various groups of children, assess the effectiveness of treatment, preventive and rehabilitation measures, prognosis of the disease, as well as determine the complex impact of chronic pathology for children [39]. Quality of life is a multifaceted concept that researchers use to measure people's assessment of their own condition. It covers physical, psychological and social well-being as perceived by the patient himself and allows us to quantify the impact on the listed components of diseases, injuries and treatment methods [40]. A number of scientific studies have shown that the quality of life of chronically ill people in all respects is lower than that of healthy people [41]. At the same time, there are also features of the violation of this indicator depending on the nosology, both in terms of the degree of decrease and in the violation of individual components of the quality of life [42]. In addition, it was found that the quality of life during an exacerbation of the disease significantly worsens compared to remission, and also depends on the frequency of exacerbations [43]. At the same time, it should be noted that the quality of life is not always identical to the severity and duration of the disease [44]. Thus, at the onset of the disease, all components of the quality of life can be greatly reduced as a result of stress from the fact of diagnosis, and then the patient gets used to the presence of a chronic disease and his quality of life can increase again. In addition, quality of life evaluates how exactly the patient tolerates his condition: optimistic people have higher scores than pessimists [45].
We have found that children with hypertension have a sharp limitation in physical, mental and social adaptation, and the process of interpersonal interaction of the sick child with others changes, since he often cannot attend children's groups or fully communicate with peers. Constant emotional stress leads to socio-psychological maladaptation, which significantly reduces the quality of life of patients with hypertension [46]. Traditional examination methods provide a one-sided picture of the disease and the effectiveness of treatment, but do not allow assessing the child’s psychological and social maladaptation and his attitude towards his condition [47]. Studying the quality of life of children opens up the possibility of a full-fledged comprehensive analysis of the child’s health status and increases the effectiveness of the medical care provided to him. Information about the parameters of a child’s quality of life can be valuable not only for pediatricians, psychologists and other specialists, but also for the child’s parents. Including an assessment of quality of life in a comprehensive rehabilitation program for children with hypertension can solve this problem, which will improve the quality of medical and social care for this group of patients [48].
Thus, to summarize the above, current problems for children with hypertension are improving the quality of diagnosis of this pathology by clinic doctors, timely referral of such patients to specialized medical institutions for complex treatment and subsequent regular follow-up in order to assess the effectiveness of the therapy and its timely correction in if necessary. It is extremely important to organize individual and group therapeutic training for children with hypertension in outpatient and inpatient settings with the publication of specialized literature for doctors, nurses, patients and their parents. It is necessary to unimpededly provide patients with hypertension at their place of residence with the necessary medications and individual means of self-monitoring (glucometers, test strips). Active scientific research is required aimed at developing effective pathogenetic treatment of hypertension in Russia.
Glycogenosis type I (Gierke's disease)
The clinical manifestations of glycogenosis type I in newborns, infants and older children are not the same. The reason is the differences in diet and nutrition in these age groups.
Sometimes fasting hypoglycemia occurs in the first days and weeks of life, but in most cases the disease is asymptomatic, since the infant feeds frequently and receives sufficient amounts of glucose. Often the disease is diagnosed several months after birth, when the child is found to have an enlarged abdomen and hepatomegaly. There may be shortness of breath and low-grade fever without signs of infection. Shortness of breath is caused by hypoglycemia and lactic acidosis due to insufficient glucose production. As the intervals between feedings increase and the baby begins to sleep at night, symptoms of hypoglycemia appear, especially in the morning. The severity and duration of hypoglycemia gradually increase, leading to systemic metabolic disorders.
If treatment is not carried out, the child's appearance changes. Characterized by muscle and skeletal wasting, delayed growth and physical development, and deposition of fat under the skin. The child becomes like a patient with Cushing's syndrome. The development of cognitive and social skills is not affected unless repeated episodes of hypoglycemia cause brain damage. If the child does not receive enough carbohydrates and the hypoglycemia of fasting persists, then the delay in growth and physical development becomes pronounced. Some children with glycogen storage disease type I die from pulmonary hypertension.
Platelet dysfunction is manifested by repeated nosebleeds or bleeding after dental and other surgical procedures. There are disturbances in platelet adhesion and aggregation; The release of ADP from platelets in response to adrenaline and contact with collagen is also impaired. Thrombocytopathy is caused by systemic metabolic disorders; after treatment it disappears.
Ultrasound and excretory urography reveal enlarged kidneys. In most patients, there are no significant renal dysfunctions; only an increase in GFR (glomerular filtration rate) is noted. In very severe cases, tubulopathy with glycosuria, phosphaturia, hypokalemia and aminoaciduria (as in Fanconi syndrome) may develop. Albuminuria is sometimes observed in adolescents, and young adults often develop severe kidney damage with proteinuria, increased blood pressure (blood pressure) and a decrease in creatinine clearance due to focal segmental glomerulosclerosis and interstitial fibrosis. These disorders lead to end-stage renal failure.
The spleen is not enlarged.
Without treatment, levels of free fatty acids, triglycerides, and apoprotein C-III, which is involved in the transport of triglycerides and triglyceride-rich lipoproteins, increase sharply. Phospholipid and cholesterol levels increase moderately. Very high levels of triglycerides are due to their excessive production in the liver and a decrease in their peripheral metabolism due to decreased lipoprotein lipase activity. With severe hyperlipoproteinemia, eruptive xanthomas may appear on the extensor surfaces of the limbs and buttocks.
Lack of treatment or improper treatment leads to delayed growth and sexual development.
Liver adenomas for unknown reasons occur in many patients, usually between the ages of 10 and 30 years. Adenomas can become malignant, and hemorrhages into the adenoma are possible. On liver scintigrams, adenomas appear as areas of reduced isotope accumulation. Ultrasound is used to detect adenomas. If malignant growth is suspected, MRI (magnetic resonance imaging) and CT (computed tomography) are more informative, allowing one to trace the transformation of a small, clearly demarcated tumor into a larger one with blurred edges. It is recommended to periodically measure serum alpha-fetoprotein levels (a marker for hepatocellular carcinoma).
The severity of fasting hypoglycemia decreases with age. Body weight increases faster than brain weight, so the relationship between the rate of glucose production and utilization becomes more favorable. The rate of glucose production is increased by the activity of amylo-1,6-glucosidase in the liver and muscles. As a result, fasting glucose levels gradually increase.
The clinical manifestations of glycogenosis type Ia and type Ib are the same, but with glycogenosis type Ib, persistent or transient neutropenia is observed. In severe cases, agranulocytosis develops. Neutropenia is accompanied by dysfunction of neutrophils and monocytes, therefore increasing the risk of staphylococcal infections and candidiasis. Some patients develop inflammatory bowel disease, similar to Crohn's disease.
Graves' disease
Thyrotoxicosis
10118 10 September
IMPORTANT!
The information in this section cannot be used for self-diagnosis and self-treatment.
In case of pain or other exacerbation of the disease, diagnostic tests should be prescribed only by the attending physician. To make a diagnosis and properly prescribe treatment, you should contact your doctor. Graves' disease: causes, symptoms, diagnosis and treatment methods.
Definition
Graves' disease (other names: Graves' disease, toxic goiter) is a hereditary autoimmune disease that can develop at any age. Women get sick about 5 times more often than men. The disease is characterized by excessive production of thyroid hormones, which leads to thyrotoxicosis (excess of thyroid hormones in the bloodstream).
It has been noticed that the pathological process often starts in situations where the body is under stress.
Causes of Graves' disease
Graves' disease is associated with a congenital defect of specific T-lymphocytes (in the body, T-lymphocytes regulate the speed of the protective reaction, its duration, and provide cellular immunity). Due to the breakdown of immunological tolerance, B-lymphocytes are activated, producing thyroid-stimulating immunoglobulins - AT-rTSH. AT-rTSH act on TSH receptors and begin to stimulate thyroid cells. This leads to a marked increase in the production of thyroid hormones (T4 and T3), as well as an increase in the size of the thyroid gland (hypertrophy).
The trigger for impaired immunological tolerance can be a viral infection, stress, or smoking.
In Graves' disease, the thyroid gland may become enlarged to the point that it is visually visible. If left untreated, it begins to compress the esophagus and trachea, causing problems with swallowing and breathing.
Classification of the disease
Graves' disease is classified according to the size of the goiter, as well as the severity of thyrotoxicosis syndrome.
In Russia, until now, the classification of endemic goiter according to O.V. is usually used. Nikolaev, proposed back in 1955.
0th degree – the thyroid gland is not visible and not palpable;
1st degree - the thyroid gland is not visible, but the isthmus is palpable and visible when swallowing;
2nd degree - the thyroid gland is visible during swallowing, palpated, the shape of the neck is not changed;
3rd degree – the thyroid gland is visible, the contour of the neck changes (“thick neck”);
4th degree – large goiter that disrupts the configuration of the neck;
5th degree - a huge goiter, compressing the esophagus and trachea.
Classification by severity of thyrotoxicosis
Subclinical period - the disease is established on the basis of hormonal studies; the clinical picture may be absent or erased.
Manifest period – there is a detailed clinical picture of the disease.
Complicated period - there are complications (atrial fibrillation, heart failure, thyrogenic relative adrenal insufficiency, dystrophic changes in parenchymal organs (liver, spleen, etc.), psychosis, significant body weight deficiency).
Symptoms of Graves' disease
Graves' disease has a wide variety of clinical manifestations. Patients complain of rapid heartbeat (tachycardia), arrhythmia, fever (intolerance to warm and stuffy rooms), heavy sweating, a feeling of internal trembling, weight loss with good appetite, frequent loose stools. Characterized by an increase in systolic (“upper”) pressure and a decrease in diastolic (lower) pressure. Many suffer from sleep disturbances, fatigue and decreased performance. Swelling of the legs, muscle weakness, and an unpleasant feeling of tightness in the neck are noted.
The second name of the disease – Basedow’s disease – indicates that patients often experience ophthalmological disorders (endocrine ophthalmopathy), which are manifested by bulging eyes (damage to the soft tissues of the orbit). You can hear complaints about regular lacrimation, pressure and “sand” in the eyes, double vision, swelling of the eyelids, photophobia.
Palpation examination reveals an enlarged thyroid gland in approximately 80% of patients.
Patients of reproductive age may experience a decrease in libido (in men); women are characterized by menstrual irregularities.
As a rule, symptoms develop quickly, so patients return to see a doctor within 6-12 months. after their manifestation.
Diagnosis of Graves' disease
If a patient is suspected of having thyrotoxicosis, the TSH level should be examined using a highly sensitive method.
Publications in the media
Glycogenosis is a group of hereditary diseases caused by a deficiency of one or more enzymes involved in the synthesis and breakdown of glycogen, and characterized by the accumulation of pathological amounts or types of glycogen in tissues. Symptoms occur due to the accumulation of glycogen or its intermediate metabolites or due to a lack of glycogen end products, especially glucose. Glycogen and some of the intermediate metabolites deposited in tissues can be detected by MRI. Differences in severity and age of onset of clinical manifestations are caused by the involvement of different isoenzymes or other components of damaged enzyme systems. The incidence of all forms of glycogen storage diseases is 1:40,000 population.
Genetic classification and clinical picture
• Glycogenosis type 0 (*240600, 12p12.2, GYS2 gene [138571], r) - deficiency of glycogen synthetase (EC 2.4.1.11) of the liver. Clinical picture: hypoglycemia and hyperketonemia on an empty stomach, convulsions, hyperglycemia and hyperlactatemia after meals.
• Glycogenosis type I(a) (232200, 17q21, Â) - deficiency of glucose-6-phosphatase (EC 3.1.3.9), leading to excessive accumulation of glycogen of normal chemical structure (especially in the liver and kidneys). It is observed much more often than other glycogenoses. Clinical picture: hypoglycemia, arterial hypertension, growth retardation, late puberty, abdominal enlargement, liver adenomas, hepatocellular carcinoma, hepatoblastoma, liver enlargement, chronic pancreatitis, xanthomas, arachnid hemangiomas, gouty tophi, proteinuria, hematuria, renal failure, central segmental glomerulosclerosis , uric acid kidney stones, gouty arthritis, hemorrhagic diathesis, pulmonary arterial hypertension. Laboratory data: glucose-6-phosphatase deficiency, hyperlipidemia, hyperuricemia, hyperlacticacidemia, ketonemia, metabolic acidosis. Synonyms: von Gierke disease, nephromegalic glycogenosis, von Gierke van Creveld syndrome, von Gierke van Creveld disease.
• Glycogenosis type Ib (232220, r) - mutations in the glucose 6 phosphate transporter gene (*602671, 11q23.3). Clinical picture: diarrhea, poor appetite, Crohn's disease, chronic osteomyelitis, perianal abscesses, neutropenia, hypochromic anemia, thrombocytopenia, secondary amyloidosis, proteinuria, hyperlipidemia.
• Glycogenosis type Ic (232240, r) - a defect in the glucose 6 phosphate transporter (*602671, 11q23.3). Clinical picture: hypoglycemia, arterial hypertension, proteinuria, hematuria, renal failure, central glomerulosclerosis, delayed growth and puberty, liver tumors (adenomas, hepatocellular carcinoma, hepatoblastoma), liver enlargement, chronic pancreatitis, xanthoma, cutaneous angioma, gouty tophi, gouty arthritis, normal leukocyte function, pulmonary arterial hypertension. Laboratory data: phosphate pyrophosphate translocase deficiency, hyperlipidemia, hyperuricemia, hyperlacticacidemia, ketonemia, metabolic acidosis. Drugs (allopurinol) should be prescribed with caution. The course is usually chronic and progressive.
• Glycogenosis type IIa (153360 - deficiency of lysosomal a-1,4 glucosidase, leading to excessive accumulation of glycogen of normal chemical structure in the heart, skeletal muscles, liver, brain. Clinical picture: cardiomyopathy, cardiomegaly, arterial hypotension, myotonia, muscle weakness, fatigue , enlarged tongue, death in the first year of life, respiratory failure, shortness of breath, cerebral artery aneurysms. Synonym: Pompe disease.
• Glycogenosis type IIb (300257, defect in the lysosome-associated membrane protein LAMP2, r). Clinical picture: proximal muscle weakness, hypertrophic cardiomyopathy, heart failure, AV block, mental retardation. Laboratory data: accumulation of glycogen in lysosomes; Acid maltase activity is normal. Synonyms: Antopol's disease, Danon's disease (300257, defect of the lysosomal protein LAMP2).
• Glycogenosis type III (*232400, 1p21, AGL gene, GDE, r) - deficiency of amylo-1,6-glucosidase, leading to the accumulation of glycogen of abnormal structure with short external chains in the liver and muscles. Clinical picture: myopathy, liver enlargement, hypoglycemia, ketoacidosis, muscle weakness with muscle atrophy, “angelic” face, tendency to bleeding (including nosebleeds), ventricular hypertrophy on the ECG. Synonyms: Measles disease, Forbes disease, limit dextrinosis.
• Glycogenosis type IV (*232500, 3p12, GBE1, r gene) - deficiency of the 1,4-a-glucan branching enzyme (EC 2.4.1.18), leading to the accumulation of glycogen of an abnormal structure with long chains in the liver, kidneys, muscles and others tissues. Clinical picture: liver cirrhosis, portal hypertension, liver failure, hypoglycemia, cardiomyopathy, heart failure, myopathy, pelvic-brachial muscular dystrophy, spinal hyperlordosis, waddling gait, weakness of the proximal muscles of the limbs, death before 4 years of age. Synonyms: Andersen's disease, amylopectinosis.
• Glycogenosis type V (*232600, 11q13, PYGM gene, r) is a defect of amylophosphorylase (EC 2.4.1.1), causing the accumulation of glycogen of normal chemical structure in the muscles. Clinical picture: weakness and atrophy of skeletal muscles, muscle pain during exercise, myoglobinuria. Synonyms: McArdle-Schmid-Pearson disease, myophosphorylase deficiency, McArdle disease.
• Glycogenosis type VI (*232700, 14q21–q22, PYGL gene, r) - deficiency of amylophosphorylase (EC 2.4.1.1), leading to the accumulation of glycogen of normal structure in hepatocytes and leukocytes. Clinical picture: liver enlargement, ketosis, hypoglycemia, growth retardation. Synonyms: Girsa disease, hepatophosphorylase deficiency.
• Glycogenosis type VII (*232800, 12q13.3, PFKM gene, r) - myopathies and liver enlargement caused by phosphofructokinase 6 deficiency (EC 2.7.1.11). Clinical picture: myopathy, liver enlargement, muscle weakness, cramps, hemolysis, mild polycythemia, reticulocytosis, moderate jaundice, cholelithiasis. Laboratory data: muscle phosphofructokinase deficiency, myoglobinuria, hyperuricemia. Synonyms: hepatophosphoglucomutase deficiency, Thomson's disease.
• Glycogenosis type VIII (*261750, Xp22.2 p22.1, PHKA2 gene, PHK; *311870, Xq12 q13, PHKA1 gene, À) - deficiency of phosphorylase kinase (EC 2.7.1.38) in muscles. Clinical and biochemical disorders disappear with age, and in most adult patients the disease is asymptomatic. Clinical picture: liver enlargement, growth retardation, renal tubular acidosis. Laboratory findings: hepatic phosphorylase kinase (PHK) deficiency; muscle phosphorylase kinase within normal limits; increased levels of glutamate-pyruvate and glutamate-oxalo-acetate transaminases; hypercholesterolemia; hypertriglyceridemia; ketonemia due to fasting; There is no hyperlactic acidemia or hyperuricemia. Synonyms: Tarui disease, liver and muscle phosphorylase kinase deficiency. Note: Some enzyme subunits have loci at 16p12.1 p11.2.
• Glycogenosis type VIIIb (261740, r) is an extremely rare form of phosphofructokinase deficiency (EC 2.7.1.38), limited to the heart muscle.
• Glycogenosis type VIIIc (*261750, r) - deficiency of phosphorylase kinase (EC 2.7.1.38) in the liver and muscles. Clinical picture: liver enlargement, diarrhea, growth retardation, muscle hypotonia, moderate weakness. Laboratory findings: phosphorylase kinase deficiency in liver and muscle with glycogen accumulation.
TREATMENT • For types 0, I and III - prevention of hypoglycemia and lactic acidosis by prescribing fractional doses of carbohydrates, which helps maintain normal blood glucose levels, prevent the development of lactic acidosis, hyperuricemia and hyperlipidemia. In addition, a continuous supply of high molecular weight dextrans is used through an endonasal probe. Allopurinol is prescribed for the prevention of gout and urate kidney stones • Limiting anaerobic exercise reduces muscle symptoms of types V and VII • For type VIII, limiting physical activity and drinking plenty of fluids is recommended • There are no effective treatments for other types.
ICD-10 • E74.0 Glycogen storage diseases
Newspaper "News of Medicine and Pharmacy" Gastroenterology (304) 2009 (thematic issue)
Liver pathology is caused by a heterogeneous group of hereditary diseases caused by various types of carbohydrate metabolism disorders. There are:
— disturbances in the metabolism of mono- and di-saccharides; - storage diseases - glycogenosis; — connective tissue pathology — mucopolysaccharidosis; - other.
Disorders of mono- and disaccharide metabolism
Fructosemia
Etiology and pathogenesis.
The disease is caused by a congenital absence of the enzymes fructose phosphate aldolase and fructose diphosphate aldolase. Excessive accumulation of fructose phosphate impairs glycogenolysis, leading to hypoglycemia. The liver has an insufficient amount of the enzyme fructose-1-phosphate-aldolase, as a result, metabolic products (fructose-1-phosphate) accumulate in the body (liver, kidneys, intestinal mucous membranes) and have a damaging effect. Morphologically, fatty infiltration and moderate perilobular fibrosis are revealed in the liver.
Clinical picture.
Symptoms occur when introducing sweet foods or fruit juices into the diet, i.e. products containing fructose. From the 2nd to 4th month, dyspeptic symptoms and conditions of acute hypoglycemia develop, which are manifested by pallor, lethargy, sweating, and the smell of acetone. In severe cases, hypoglycemic coma may develop with loss of consciousness and convulsions. It is typical that hypoglycemia occurs after eating. As children grow older, they themselves refuse sweet foods. A consistent feature is hepatomegaly (usually with enlargement of both lobes) with a smooth margin and some thickening of the liver.
Treatment
. A diet devoid of fructose, the main sources of which are honey, sugar cane and beets, fruits, jams, marmalade, carrots, cocoa, chicory, turnips. Patients are allowed to consume milk and dairy products, eggs, olives, sunflower oil, and animal fats. Women's and cow's milk are allowed. Powdered milk should be without sugar (sucrose). All types of cheeses and natural fermented milk products (unsweetened) are allowed. Milk with added sucrose, condensed milk, and sweetened fermented milk products are prohibited. Meat and fish are allowed. Sausages and sausage products, canned food are excluded. Fats are included in the diet with virtually no restrictions (butter and vegetable oil, margarine). Almost all fruits are prohibited. You can eat lemons and chestnuts. Vegetables allowed are green beans, watercress, lettuce, leeks, cabbage, and spinach. Allowed are natural wheat or rye flour, rice, bread, semolina, tea, coffee, cocoa without sugar, glucose, maltose, dextrin-maltose, saccharin. Soy, flour with sucrose, biscuits, cakes, lemonade and all carbonated fruit drinks, juices, syrups, sugar, jam, nougat, sweets, all medications containing sugar, sorbitol (granules, dragees, powders, pills) are prohibited.
Newborns are prescribed sugar-free milk formulas. Children of the first year receive milk formulas containing only lactose and dextrin-maltose. Instead of fruit purees and juices, food is supplemented with glucose (from 30 to 60 g). When introducing complementary foods earlier than healthy children, meat, fish, cheese, and eggs are prescribed. Ascorbic acid is used without sugar. Dietary nutrition is indicated up to 5–6 years of age; only after reaching this age can foods from the prohibited list be included in the child’s diet in limited quantities, under the supervision of a doctor and biochemical blood tests.
Galactosemia
Frequency 1: 35,000–50,000 population.
Etiology and pathogenesis
.
Hereditary enzymopathy. Inherited in a recessive manner. Galactosemia (Fig. 1) is based on a violation of galactose metabolism due to the absence of the enzyme galactose phosphate uridyl transferase. As a result, galactose and galactose phosphate accumulate in the blood in high concentrations. There is a disruption in the process of enzymatic conversion of galactose into glucose with the accumulation of galactose and its metabolic products in cells, which has a damaging effect on the functions of the liver, brain, eye lens, and kidneys. Clinical picture
. Clinical signs of the disease appear early - 1–2 weeks after the birth of the child. Appetite disappears, lethargy, vomiting, and diarrhea appear. There is a lack of body weight. Hepato- and splenomegaly gradually develop, and persistent hyperbilirubinemia appears, mainly due to direct bilirubin. Cataracts leading to blindness are common. There may be symptoms indicating damage to the kidneys (proteinuria, hyperaminoaciduria), the central nervous system (delayed psychophysical development). After a tea-water break, the condition improves, but the introduction of milk causes a recurrence of disorders of the gastrointestinal tract. If the diagnosis is not made in a timely manner, the disease progresses, leading to severe consequences or death.
Diagnostics:
1. Determination of the concentration of galactose-1-phosphate in erythrocytes (increased). 2. Study of the activity of galactose-1-phosphate-uridyltransferase in erythrocytes. 3. Increasing the level of galactose in the blood and urine (by chromatography). 4. Guthrie microbiological test.
Treatment.
Diet therapy is the only treatment method. Lactose-free formulas are used to feed the baby. Whole milk products are excluded from the diet of older children.
Glycogenoses
Etiology and pathogenesis
. A group of hereditary diseases of polysaccharide metabolism that develop as a result of impaired synthesis or breakdown of glycogen into simple sugars. In this case, normal and abnormal glycogen simultaneously accumulate in liver cells and other organs and tissues.
Glycogen is a highly branched polymer of glucose in which the majority of residues have 1,4-linkages and 7–10% of the residues have 1,6-linkages. The tree-like structure undergoes superstructure and detachment of residues at the periphery of the molecule. The molecular weight of glycogen is several million; its molecules can aggregate to form structures visible under electron microscopy. Liver glycogen typically contains less than 70 mg/g and muscle glycogen less than 15 mg/g, but these values fluctuate depending on nutrition and hormonal influences. Disturbances in the structure of glycogen can be associated with both a decrease and an increase in the branching of the molecule.
The metabolic pathways for the synthesis and breakdown of glycogen in different tissues are different; for example, some reactions occur actively in the liver but are less active or absent in muscle, and a number of enzymes in muscle and liver are encoded by different genes. Plasma glucose enters the cell and is phosphorylated by glucokinase or hexokinase. The first is contained in the liver, which phosphorylates the bulk of glucose, while numerous hexokinases are more widely distributed throughout the tissues. Glucose-6-phosphate (G-6-P) is converted to glucose-1-phosphate (G-1-P) in a reversible reaction catalyzed by phosphoglucomutase. Uridine diphosphate glucose (UDPG) is synthesized from G-1-P and uridine triphosphate by the action of UDPG pyrophosphorylase. Genetic deficiency of any of these liver enzymes has not been reported. The glycogen molecule is then extended by the addition of individual glucose residues from UDPG, resulting in the formation of a polymer. This reaction is catalyzed by glycogen synthase, which exists in active dephosphorylated and inactive phosphorylated forms. The synthesis of a normally branched glycogen molecule also requires the participation of a branching enzyme (1,4-a-glycan: 1,4-a-glycan-6-glycosyltransferase), which converts the 1,4-linkage of the oligosaccharide to position 1, 6-connections.
Glucose is mobilized from glycogen by a number of enzymatic reactions. The active form of phosphorylase, phosphorylase a, directly acts on glycogen, splitting off individual glucose residues and forming G-1-P. In liver and muscle, phosphorylase is encoded by different gene products. In these tissues, the enzyme can exist in active phosphorylated and inactive dephosphorylated forms. Phosphorylase is a dimer consisting of identical subunits, and both forms of the enzyme are subject to complex allosteric regulation. Inactive phosphorylase b is converted to an active form by phosphorylase b-kinase, which exists in both active phosphorylated and inactive dephosphorylated forms. This enzyme consists of four different subunits (a, b, g, d4), and the d-chain is identical to the calcium-binding protein calmodulin. The rate of glucose mobilization by this system is regulated by a cascade of kinase reactions involving cAMP.
Based on the nature of enzyme deficiency, it is customary to distinguish 12 types of glycogenosis, among which are liver (glycogenosis types 0, I, III, IV, VI, VIII, IX, X, XI) or muscle forms (glycogenosis types V and VII). Type II glycogenosis is manifested by damage only to muscles or damage to many systems and organs (generalized form). Combinations of several types are also possible.
Clinical picture
glycogenosis is characterized by hypoglycemia (vomiting, convulsions, loss of consciousness, coma). The course of the disease depends on the location of glycogen deposition: liver, kidneys, muscle tissue. Accordingly, liver cirrhosis, renal form, and muscle syndrome (including cardiac form) are distinguished. The predominance of hypoglycemia symptoms in a newborn child can lead to sudden death syndrome. The prognosis depends on the type of disease.
The classification is based on differences in enzyme defects underlying the diseases.
Glycogenosis type 0
(aglyconosis) is characterized by a sharp decrease in glycogen reserves in the liver, a serious condition is observed up to the development of coma (hypoglycemic syndrome). Coma can occur after birth when the baby is put to the mother's breast late, and later in the morning on an empty stomach and in between feedings. If the child is left untreated, psychomotor development is impaired.
Glycogenosis type I
(Gierke's disease) (Fig. 2) - glycogenosis caused by deficiency of glucose-
6-phosphatase, leading to the inability to convert glucose-6-phosphate into glucose, which is accompanied by the accumulation of glycogen in the liver and kidneys;
inherited in an autosomal recessive manner. Enzyme defect in the liver, kidneys, and small intestinal mucosa. At its first manifestations, there is a lack of appetite, vomiting, respiratory distress syndrome, hypoglycemic convulsions (coma), which are detected immediately after birth or in infancy. Hepatomegaly and nephromegaly progress due to glycogen infiltration. Over time, the following appear: stunted growth, body disproportion (large head, short neck and legs), doll-like face, muscle hypotonia; puberty is delayed. Neuropsychic development is satisfactory. Due to severe hypoglycemia, patients are forced to constantly eat. Glycogenosis type II
(Pompe disease) - inherited in an autosomal recessive manner. Symptoms appear in the first weeks of life - up to six months after birth. The enzyme defect was found in the liver, kidneys, spleen, muscles, nervous tissue, and leukocytes. There is respiratory distress, anxiety or adynamia. Lack of appetite, growth retardation, and muscle hypotension are noted. The size of the heart, liver, kidneys, and spleen increases. The heart takes on a spherical shape, and due to myocardial hypertrophy, ECG changes appear. Hypostatic pneumonia, bronchitis, and pulmonary atelectasis often occur, myodystrophy, hyporeflexia, and spastic paralysis are observed. The muscular form of glycogenosis type II occurs only in muscles with a deficiency of acid α-1,4-glucosidase. The disease manifests itself at a later date and the clinical picture resembles myopathy.
Glycogenosis type III
(Measles disease, Forbes disease, limit dextrinosis) - glycogenosis caused by deficiency of the enzyme amylo-1,6-glucosidase. This enzyme catalyzes the cleavage of [...COC...] bonds in the glycogen molecule at branch points. The disease is accompanied by the deposition of atypical glycogen in the liver, heart, and muscles. Inherited in an autosomal recessive manner. The enzyme defect was found in the liver, muscles, leukocytes, and erythrocytes. From the first months of a child’s life, hepatomegaly, muscle hypotonia, and hypertrophy of individual muscle groups are observed. In some cases, patients experience impaired cardiac conduction and circulation, and myocardial hypertrophy. The development of the disease slows down after the age of five or during puberty.
Glycogenosis type IV
(Andersen's disease, amylopectinosis, diffuse glycogenosis with liver cirrhosis) - glycogenosis, familial liver cirrhosis, caused by a defect in the enzyme amylo-(1,4-1,6)-transglucosidase. This enzyme catalyzes the conversion of 1,4-bonds in the glycogen molecule into 1,6-bonds, that is, it causes the branching of the polysaccharide molecule. The disease is accompanied by excessive accumulation of atypical glycogen in the liver. Inherited in an autosomal recessive or sex-linked manner. An enzyme defect was found in the liver, kidneys, muscles, and leukocytes. The disease is observed from the first months of life and is characterized by hepatosplenomegaly, the development of liver cirrhosis, jaundice, and hypoglycemia.
Glycogenosis type V
(McArdle's disease, myophosphorylase deficiency) - glycogenosis associated with a defect in muscle phosphorylase. A disease caused by a violation of the catalytic function of this enzyme; accompanied by the deposition of a significant amount of glycogen in the muscles. Inherited in an autosomal recessive manner. An enzyme defect found in the muscles. Due to glycogen infiltration, skeletal muscles increase in volume and become very dense. Muscle weakness, muscle spasms, tachycardia during physical activity appear in the first ten years of life and progress. Transient myoglobinuria is observed. The concentration of lactate in the blood decreases after exercise. Males are more often (5 times) affected.
Glycogenosis type VI
(Hers disease, hepatophosphorylase deficiency) - glycogenosis caused by liver phosphorylase deficiency. Liver phosphorylase catalyzes the phosphorylation of glycogen to form glucose-1-phosphate. Disruption of this mechanism leads to excessive glycogen deposition in the liver. It is presumably inherited in an autosomal recessive manner. It usually appears in the first year of life. Characterized by significant enlargement of the liver as a result of glycogen infiltration of hepatocytes, growth retardation, doll-like face, hyperlipidemia, hyperglycemia after intravenous administration of galactose, increased glycogen content in erythrocytes. Inherited in an autosomal recessive manner. An enzyme defect was found in the liver and leukocytes. It usually appears in the first year of life.
Glycogenosis type VII
(Tarui disease, myophosphofructokinase deficiency) - symptoms are similar to type V glycogenosis. An enzyme defect has been found in muscles and red blood cells. Also characteristic are muscle weakness, fatigue, and the absence of hyperlactic acidemia after exercise.
Glycogenosis type VIII
(Thomson's disease) - rare, inheritance has not been established. An enzyme defect was found in the liver and in the brain. After birth, the size of the liver increases, nystagmus (“dancing eyes”) and ataxia appear. Neurological symptoms are progressing.
Glycogenosis type IX
(Hag's disease) - inherited in a recessive, sex-linked manner. An enzyme defect was found in the liver. Patients have hepatomegaly.
Glycogenosis type X
— the case is known in a single patient, inheritance has not been established. An enzyme defect was found in the liver and muscles. Hepatomegaly was observed, and 6 years after the onset of the disease, muscle pain and muscle spasms appeared after exercise.
Glycogenosis type XI
(Fanconi-Bickel disease) - inheritance has not been established. An enzyme defect was found in the liver and kidneys. It is characterized by a significant enlargement of the liver and a sharp retardation of growth. Symptoms of hypophosphatemic rickets are observed. During puberty, a decrease in liver size, accelerated growth, and normalization of phosphorus levels in the blood are possible.
Treatment
glycogenosis is mainly symptomatic and is aimed at changing metabolic disorders. The goal of treatment is to prevent severe hypoglycemia. Prescribe a diet rich in proteins and carbohydrates. Eating should be frequent (every 4 hours). Proteins serve as a source of amino acids - substrates of gluconeogenesis; they reduce the carbohydrate load, leading to hyperglycemia and lactic acidosis. This diet prevents fasting hypoglycemia and ketoacidosis, reduces postprandial hyperglycemia and lactic acidosis, and promotes growth. In muscular forms of glycogenosis, improvement is noted by following a diet high in protein, prescribing fructose, multivitamins, and ATP. Sometimes it is necessary to use glucagon, anabolic hormones, and glucocorticoids. Attempts are being made to administer missing enzymes to patients.
Prevention
not developed.
Mucopolysaccharidoses
Etiology and pathogenesis
. Mucopolysaccharidoses (MPS) are a heterogeneous group of diseases classified as hereditary diseases of complex sugar metabolism. MPS are accompanied by excessive accumulation in tissues and increased excretion of glycose aminoglycans (GAGs) - acidic mucopolysaccharides combined with protein and consisting of uronic acids, aminosucroses and neutral sugars. These complexes exist in the form of proteoglycans, which are the most important components of the main structural protein of hair (O-keratin) and the structural protein of connective tissue (collagen).
Most MPS are characterized by an autosomal recessive mode of inheritance, except for Hunter syndrome (X-linked recessive).
Clinical picture
. Manifestation of the disease, usually before the age of 7 years, growth retardation (up to dwarfism), joint contracture, kyphosis/kyphoscoliosis/scoliosis, massive skull with a deep and elongated sella turcica, short neck, chest deformation, paddle-shaped ribs, shortening of tubular bones , coarse facial features, corneal clouding, hepatosplenomegaly, mental retardation, convulsions, deafness, hernias (umbilical, inguinal, inguinal-scrotal), congenital heart defects, glycosaminoglycanuria (100–200 mg per day). The signs of dysmorphism characteristic of MPS are called the “gargoyl phenotype” (gargoylism is a synonym for MPS).
Among MPS, a number of types are distinguished, each of which is caused by a deficiency of a specific lysosomal hydrolase involved in the sequential cleavage of GAGs.
Type I - Hurler syndrome (4p16 - IDUA). There are subtypes: Gurler, Sheye, mixed version. All are characterized by a decrease in alpha-iduronidase activity and accumulation of dermatan and heparan sulfates in the tissues.
Type II - Hunter (Gunter) syndrome (Xq28 - IDS). Reduced activity of L-idurosulfate sulfatase and deposition of dermatan and heparan sulfates in tissues. Clinical signs are less pronounced, life expectancy is longer than with other types of MPS.
Type III - Sanfilippo syndrome (12q13.4). Depending on the nature of the primary biochemical defect, four subtypes are distinguished: A, B, C, D. The clinical picture is dominated by mental disorders: dementia, aggressiveness. Life expectancy does not exceed 20 years.
Type IV - Morquio syndrome (16q24.3 - GALNS). There are subtypes A and B. Keratan sulfate is deposited in the tissues. Skeletal lesions and disproportionately short stature predominate.
Type V - Scheie syndrome (see type I).
Type VI - Maroteau-Lamy syndrome (5q11.2 - ARSB). Deficiency of the enzyme arylsulfatase B. Dermatan sulfate accumulates in the tissues. Phenotypically resembles MPS type I, but intelligence is not reduced.
Type VII - Sly syndrome (7q21.11 - GUSB). Deficiency of the enzyme δ-glucuronidase. Dermatan, heparan and chondroitin sulfates accumulate in tissues. Phenotypically resembles type I MPS, but has a more benign course.
Diagnosis of MPS
is based on a combination of data from genealogical analysis, clinical manifestations, typical radiological data, urinary excretion of hydroxyproline (decrease), GAGs and their fractions (5-10 times higher). Accurate identification of MPS types is only possible by determining the activity of lysosomal hydrolases in blood lymphocytes and leukocytes, cultured skin fibroblasts, liver biopsies, and also in urine.
Treatment
In patients with MPS, the treatment is mainly symptomatic and consists of prescribing therapy that promotes normalization or stabilization of the pathological process in the musculoskeletal system, cardiovascular and central nervous systems, parenchymal organs, organs of vision and hearing. Plasmapheresis is considered promising.