The type of inheritance of cystic fibrosis is autosomal recessive. If the mother and father are carriers of the gene for this disease, but are not sick themselves, then the chances of having a child with cystic fibrosis are 25%. Cystic fibrosis manifests itself in the first two years of a baby’s life. Moreover, the earlier the symptoms of cystic fibrosis appear, the more severe the course of the disease and the worse the prognosis. The overall life expectancy of people with cystic fibrosis usually does not exceed 40 years.
Scheme of X-linked recessive inheritance of diseases
Causes of cystic fibrosis
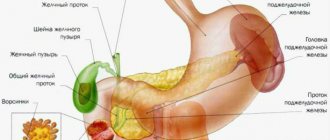
The main causes of cystic fibrosis are mutations in genes encoding a protein that takes part in the water-electrolyte metabolism of epithelial cells. As a result, the secretion they secrete becomes thick and is removed outside with great difficulty. Due to its constant accumulation, the glands undergo deformation and irreversible processes begin to occur in them. As a result, everything ends with the death of the glands and their replacement with connective tissue. Such changes lead to the organ ceasing to function.
Mucus begins to stagnate in the bronchi. This makes it difficult for the ciliated epithelium to work. Dust, small smoke particles, viruses and bacteria settle on the surface of the mucous membrane. In conditions of high humidity and temperature, microorganisms multiply well, causing an inflammatory process. Prolonged, low-grade inflammation leads to a decrease in local immunity, deformation of the ciliated epithelium of the bronchi, and even greater disruption of its function. The bronchi become less elastic, their lumen constantly narrows, creating an even more favorable environment for the proliferation of bacteria.
Constant accumulation of mucus makes breathing difficult. The body does not receive enough oxygen. As a result, the functioning of all organs and systems is disrupted.
Since the pancreas produces too thick a secretion, its ducts become clogged. This further complicates the outflow of secretions and causes inflammation. In conditions of the inflammatory process, the supply of oxygen and nutrients deteriorates. Sclerotic processes are activated in the organ. The gland becomes deformed and its function is further impaired. Since an insufficient amount of enzymes is released into the child’s duodenum, the stool becomes very viscous, constipation and bloating develop. Due to malabsorption of nutrients, the child lags behind in physical development.
Clinical manifestations of pathology
Damage to internal organs occurs due to malfunction of the exocrine glands:
- pancreas;
- salivary;
- bronchopulmonary;
- biliary;
- sweat, etc.
Instead of the usual secretion, the glands produce a thick, viscous mass, which causes constant blockage of the ducts in individual organs. As a result, some areas of the organs become blocked. After some time, the cells of the blocked area die and are replaced by fibrous tissue. Gradually, more and more new areas are “switched off”, which is why the functions of the affected organs decrease.
The first symptoms of cystic fibrosis can appear in early childhood, and the earlier they become obvious, the faster the process of accumulation of pathological changes occurs, which means the more severe the disease. The child suffers:
- chronic recurrent diseases of the respiratory organs;
- chronic bronchitis;
- chronic sinusitis, the course of which cannot be treated;
- nasal polyps;
- recurrent pancreatitis;
- respiratory failure;
- is not physically developed enough compared to his peers.
Thickening of mucous secretion occurs due to disturbances in protein synthesis, which is involved in the regulation of water-electrolyte cellular metabolism. This process is most important for the functioning of the respiratory tract, digestive tract, pancreas and liver, and subsequently the reproductive system.
Pathological changes in the bronchi and lungs are present in 95% of patients. About a third of patients suffer from intestinal secretion disorders, which manifests itself in rectal prolapse. After prescribing digestive enzymes to such patients, intestinal function returns to normal within two months.
Many children suffering from cystic fibrosis develop cough, asthma attacks and some other manifestations already in the first months of life. Sinusitis, chronic cholecystitis, and colitis syndrome are quite typical.
Signs and symptoms of cystic fibrosis
Clinical symptoms of cystic fibrosis depend on the form of the disease:
- pulmonary form. As the name suggests, this form of cystic fibrosis affects the respiratory system. The patient develops chronic bronchitis with difficult to separate sputum, and frequent pneumonia is observed. The cough is constant, exhausting, and causes insomnia. As a result, pulmonary and then heart failure develops first;
- intestinal form. It manifests itself as symptoms of gastrointestinal damage, which leads to anemia, hypovitaminosis and weight deficiency. If the liver is affected, jaundice may develop;
- mixed form. Combines the symptoms of intestinal and pulmonary forms of the disease;
- atypical forms. Meconium ileus in newborns. Viscous intestinal contents clog the newborn's intestines, which makes it impossible to pass stool.
The layering of infection is accompanied by a pronounced intoxication syndrome. The patient's temperature rises, complaints of general weakness, sweating, and severe malaise arise. The cough intensifies and often becomes debilitating. The patient loses his appetite, and his performance is greatly reduced. Bronchitis and pneumonia are difficult to treat and tend to be protracted.
In infants, the symptoms of cystic fibrosis are:
- thick and greasy feces, with a pungent, foul odor;
- rectal prolapse; enlarged liver;
- delayed physical development;
- bloating;
- deformation of the chest and phalanges of the fingers;
- dry skin;
- lingering dry cough.
Often, when kissing, a mother notices the salty taste of the baby’s skin. With additional examination, the doctor confirms the diagnosis of cystic fibrosis.
Signs and symptoms of cystic fibrosis
Cystic fibrosis from a general practitioner's perspective
| Doctors traditionally associate CF primarily with pulmonary pathology, but it is a systemic disease that affects the gastrointestinal tract and other vital organs |
In medical practice, CF is traditionally associated primarily with pulmonary pathology, but it is a systemic disease that affects the gastrointestinal tract, secondarily the heart, and other vital organs. And although the prognosis remains unfavorable and not all patients survive the age of 25, CF can be considered as a multidisciplinary clinical problem, primarily at the intersection of pediatrics and therapy
Cystic fibrosis (CF) is a genetic autosomal recessive (a/r) monogenic disease caused by a mutation in the CF transmembrane regulator (CFTR) gene. It is characterized by a violation of the secretion of the exocrine glands of vital organs with damage primarily to the respiratory and gastrointestinal tract, severe course and poor prognosis [1, 2, 3]. It was first isolated from the celiac disease group in 1936 by Viennese pediatrician Guido Fanconi [9].
The initial mutation most likely arose somewhere on the border of modern Holland and Germany. The gene is detected, according to European data, with a frequency of 1 case per 1500 people, second in prevalence only to trisomy syndrome [12]. Gradually spreading to the east, where the incidence is clearly increasing both as a result of improved diagnostic measures and as a true indicator. The probability of having a sick child, according to European data, is 1:2000 - 1:2500 live births [13]. The number of diagnosed patients in developed countries is 7-8 per 100 thousand population, of which the number of patients over 18 years of age is 20%, and in the USA even 32% [8]. In Russia, most of these patients are not detected or are detected late, often in an advanced stage of the disease, and therefore the prevalence of CF, according to our statistics, does not exceed 1:100,000, but according to calculations by the Medical Genetics Center of the Russian Academy of Medical Sciences, it should be 1:12,000 newborns [4 ].
After its first descriptions, CF was considered a fatal disease, since most children did not survive the five-year mark. Until the 1980s, 80% of patients were under 20 years of age [13]. But even in modern conditions, there is a wide variation in the life expectancy of patients, which is due to the level of development of society, awareness of the urgency of the problem, and the degree of organization of specialized centers. In the UK, USA, Australia, newborns with CF are guaranteed 40 years of life, in other developed countries - an average of 31, in Latin America - 10 years (95% of CF patients are not detected here), in Russia, according to the results of the work of the CF center based on the republican children's hospital (Moscow), - 16 years. Treatment of CF requires large financial investments. So, in the USA they amount to 15 thousand dollars a year, in the UK - 11 thousand pounds. Art., in the Russian CF Center - $10,500 per year [2].
Thus, CF is an important medical and social problem, which is due not only to the large moral, physical and material costs of the family, practical healthcare authorities and society as a whole for the diagnosis, treatment and social adaptation of patients, but also to the achievements of scientific and practical medicine, which have made it possible to increase life expectancy of patients.
The CF gene is localized on chromosome 7 (7q31). A/R is transmitted with varying degrees of expressiveness of characteristics. One gene can have many mutations, each of which is characteristic of a specific population or region. The main mutation delta-F508, i.e. a substitution of the amino acid phenylalanine at position 508, occurs in 56% of cases in Russia, and in 41% of cases in Moscow. The frequency of other mutations - W1282X, N1303K, etc. - is 3-5% [4, 5, 11]. Severe CF is characteristic of homozygotes for delta-F508, as well as with mutations W1282X, G542X, N1303K, both in the homozygous state and in combination with delta-F508. A relatively mild course of CF is observed in children with mutations R334W, S1196X (even in combination with delta-F508). But it is difficult to predict the course and prognosis of the disease even with the same mutation. Sometimes, with early diagnosis of CF and adequate treatment, irreversible changes in vital organs quickly develop. At the same time, in some patients, even with late diagnosis and, accordingly, late treatment, a favorable course of CF was observed, allowing them to reach adulthood and sometimes adulthood. Such clinical polymorphism can be explained by the genetic characteristics of the child himself, as well as the nature of life, immune reactions, etc.
The pathogenesis of CF is associated with a defect in the synthesis of protein, which acts as a chloride channel involved in the water-electrolyte metabolism of epithelial cells of the respiratory tract, gastrointestinal tract, pancreas, liver, and reproductive system. In 70% of mutations in the European population, there is a loss of the amino acid phenylalanine in the protein. Due to the inability of the defective protein to adequately perform the work of the chloride channel, Cl– ions accumulate inside the cell. The electrical potential in the lumen of the excretory ducts changes, and sodium ion rushes into the cell. The latter, in turn, acts as a pump, which causes increased absorption of water from the pericellular space [7]. As a result, the secretion of most of the exocrine glands thickens, its evacuation becomes difficult, and secondary changes occur in the organs, the most serious in the bronchopulmonary system. Chronic inflammation of varying severity develops in the walls of the bronchial tree, the connective tissue framework is destroyed, and bronchiolo- and bronchiectasis are formed. In conditions of constant obstruction by viscous sputum and progressive destruction of the pulmonary parenchyma, bronchiectasis becomes common, hypoxia increases, pulmonary hypertension and the so-called “pulmonary heart” develop. The pathogenesis of CF includes other mechanisms. In particular, there is a decrease in IgA production, a decrease in antiviral immunity, the formation of interferon, and the phagocytic activity of leukocytes, especially their microbicidal effect [1]. The alveolar macrophage is the main phagocyte of the lungs [7] and is the main source of interleukin 8 (IL8), the leading chemoattractant for neutrophils. In CF patients, the concentration of IL8 in sputum and bronchoalveolar fluid increases, which positively correlates with the severity of the bronchopulmonary process [10, 15]. At the same time, other chemoattractants (C5a, leukotriene B4), cytokines (IL1, IL6, tumor necrosis factor alpha), and elastase, which play an important role in the formation of chronic inflammation, are found in large quantities in CF [15]. Many neutrophils attracted to the respiratory tract increase the amount of purulent sputum. Destroying neutrophils release DNA, which also increases the viscosity of sputum. Endo- and panbronchitis is preceded by a viral infection of the nasopharynx, larynx and bronchi, leading to the death of ciliated epithelial cells and opening the way for microbial flora (especially against the background of JgA deficiency). Bacteria, especially Pseudomonas aeruginosa, and endogenous proteases, primarily elastase, appear as damaging agents. Protective factors against the latter are alpha-one antitrypsin and secretory leukoprotease inhibitor, but in CF patients they are suppressed by the huge amount of neutrophil protease. This allows proteases to directly destroy the epithelium and framework of the bronchial tree, further altering mucociliary clearance. In the early stages of CF, the microbial landscape of the bronchi is dominated by staphylococcus, later by Pseudomonas aeruginosa, which is explained by the ability of the pathological CF protein to change the conditions of formation and the quantitative composition of “sugars” on the surface of the epithelial cell of the respiratory tract. This facilitates the adhesion of microbes, especially Pseudomonas aeruginosa [11, 15, 16].
Bronchopulmonary changes predominate in the clinical picture and determine the prognosis in 95% of patients [13, 16]. Doctors traditionally associate CF primarily with lung pathology, but it is a systemic disease that affects the gastrointestinal tract, secondarily the heart, and other vital organs. And although the prognosis remains unfavorable and not all patients survive the age of 25, CF can be considered as a multidisciplinary clinical problem, primarily at the intersection of pediatrics and therapy.
Patients with cystic fibrosis and their families should be followed up in specialized centers. But in our country there are few such centers, and given the distances and the lack of necessary funds for travel expenses, they become completely inaccessible to patients. Therefore, the principles of managing patients with CF should be familiar to doctors in a wide medical network.
- Clinical picture
The pronounced polymorphism of CF determines different variants of its course. In 8-20% of homozygous cases, CF manifests itself from birth as meconium ileus with a possible outcome in meconium peritonitis. In less tragic situations, the stool is copious, with a strong disgusting odor, “greasy”. In 1/3 of patients, rectal prolapse is observed, but when an adequate dose of modern digestive enzymes is prescribed, this complication spontaneously resolves after 1.5-2 months. It should be recalled that meconium ileus, as a rule, indicates the presence of CF in a newborn (differential diagnosis - DD - is carried out primarily [14] with familial chloride diarrhea syndrome). In school-age patients, the first symptoms may be “intestinal colic.” When examining such children, “dense formations” (feces mixed with thick dense mucus) are palpated in the abdominal cavity, which cause bloating, repeated vomiting, and constipation. Distal intestinal obstruction or the equivalent of meconium ileus in its acute, subacute or chronic form can be observed in adolescents, young men and even adults [11, 14, 15], necessitating DD with visceral steal syndromes, superior mesenteric artery, polyposis, congenital and acquired intestinal pseudo-obstruction.
After the administration of enzymes, intestinal symptoms are relegated to the background, giving way to pulmonary symptoms. Acute onset of bronchopulmonary pathology is observed rarely. Usually chronic (in 1/3 of cases - obstructive) bronchitis gradually develops. At birth, the respiratory tract is intact, but already in the neonatal period and infancy, coughing, attacks of suffocation, shortness of breath, sometimes vomiting, and recurrent bronchopneumonia occur. Then emphysema forms, often atelectasis, and bronchiectasis quickly appears. Periodically, a painful, severe cough occurs, especially at night. The sputum is viscous, sometimes purulent. There may be symptoms of bronchial asthma. Initially, the auscultatory picture outside of exacerbations remains unchanged, but a careful examination reveals slight shortness of breath, an increase in chest volume mainly due to the anteroposterior size, and a slight but constant decrease in chest excursion. As a result of overload of the small circle, a “pulmonary heart” develops. Chronic hypoxia leads to deformation of the terminal phalanges of the fingers like “drumsticks” and nails like “watch glasses.”
Damage to the pancreas causes malabsorption syndrome with dystrophy and copious fatty stools. In the later stages, diabetes mellitus develops and, in 13% of patients with mixed and intestinal forms of CF, liver cirrhosis develops. Liver cirrhosis is typical for the W1282X, delta-F508 and X1303K mutations. In 5-10% of patients, biliary cirrhosis with portal hypertension is detected. In general, clinical, laboratory, instrumental and morphological changes in the liver are detected in 86% of patients [2, 15, 16].
Since all organs containing mucus-producing glands are affected, colitis syndrome, chronic cholecystitis, and sinusitis are typical.
- Diagnosis and principles of observation [1, 12, 16]
1. General symptoms: dystrophy, retarded physical development, recurrent chronic respiratory diseases, nasal polyps, persistent chronic sinusitis, chronic bronchitis, recurrent pancreatitis, malabsorption syndrome with “fatty stools,” “unclear” dyspeptic disorders, liver cirrhosis, reduced fertility in women, respiratory failure, “thermal collapse.” Chronic colitis, cholecystitis in relatives.
2. Sweat test: iontophoresis with pilocarpine. An increase in Cl more than 60 mmol/l is a probable diagnosis; Cl concentration > 100 mmol/l is a reliable diagnosis. In this case, the difference in the concentration of chlorine and sodium should not exceed 8-10 mmol/l. The sweat test must be positive at least three times to make a definitive diagnosis. A sweat test should be performed on every child with a chronic cough.
3. Chymotrypsin in stool [6]. Chymotrypsins are a group of endopeptidases produced by pancreatic cells as chymotrypsinogens A, B and C. After entering the gastrointestinal tract, they are activated. To determine chymotrypsin in stool, it is necessary to stop taking digestive enzymes at least 3 days before the test. In CF, the concentration of chymotrypsin in feces is reduced. (The sample is not standardized. Standard values are developed in a specific laboratory.) Differential diagnosis is carried out with exocrine pancreatic insufficiency of any other etiology, Shwachman syndrome, condition after gastrectomy according to Billroth II, protein deficiency.
False-negative values are obtained with a slight to moderate decrease in the exocrine function of the pancreas.
False-positive values may occur if enzyme preparations are discontinued late before the test.
4. Determination of fatty acids in stool [6]. Normal is less than 20 mmol/day. Borderline values are 20-25 mmol/day. The test is positive when exocrine function decreases by at least 75%. Differential diagnosis:
- deficiency of conjugated fatty acids in the small intestine (as a condition for the emulsification of fats with their subsequent fermentation by lipase) with liver failure, bile duct obstruction, bacterial contamination of the small intestine with increased hydrolysis of bile acids;
- ileitis;
- malabsorption in celiac disease;
- enteritis;
- intestinal lymphomas;
- Whipple's disease;
- food allergies;
- accelerated passage of food during diarrhea, carcinoid syndrome, hyperthyroidism.
5. DNA diagnostics. The most sensitive and specific. False results are obtained in 0.5-3% of cases. Justified for countries where the frequency of delta-F508 is above 80%. In Russia, due to the relatively low frequency of the latter, DNA diagnostics is difficult and expensive. As noted above, the frequency of different mutations varies among ethnic groups. It is recognized that if none of the ten “local” mutations is found on one or another allele of a child’s chromosome and his parents are not relatives, the likelihood of CF in this patient is negligible. DNA testing of parents and other family members without clinical signs of the disease is not advisable, except in cases of prenatal diagnosis.
6. Prenatal DNA diagnostics. Study of intestinal alkaline phosphatase isoenzymes from amniotic fluid. Possibly from 18-20 weeks of pregnancy. False positive and false negative values are obtained in 4% of cases.
7. Currently, in our country, about 75% of mutant chromosomes are identified, which does not allow mass screening . But it is possible to conduct cascade screening [2], when the family of the CF patient and relatives are in the center. Screening of newborns can be carried out using the IRT (immunoreactive trypsin) method or the VM-labstick test (Hoechst). The IRT method is relatively expensive and produces about 10% false positive and false negative results. The VM test is significantly cheaper, but can produce 15% false negative results. Newborn screening seems to be a promising area, as it allows us to determine the frequency of CF in the country, start early intensive treatment and identify couples in need of genetic counseling.
- Treatment
Treatment of CF requires a comprehensive approach. Once again it should be emphasized that it should be carried out in specialized centers and under their control. It is impossible to treat a child without close cooperation with parents. Therefore, parents should be informed about the essence of the disease, the nature and characteristics of the process, treatment methods, and trained in the diagnosis of deterioration, a number of treatment and rehabilitation methods. In our country, children with CF have benefits, but their list should be significantly expanded and extended in terms of age. Visits to the doctor should occur at least once every 3 months. Anthropometric data, external respiratory function, general blood and urine tests, coprogram, sputum analysis for flora and its sensitivity to antibiotics are assessed. According to indications, a chest X-ray, echography of the liver and heart are performed, and the immune status is examined. A “day hospital” is desirable, which is widespread in the West and well accepted by our patients and their parents [2, 13, 15, 16]. First of all, corrections are made to the treatment and rehabilitation regime. It is necessary to effectively clear the bronchial tree of viscous sputum, fight infection and ensure good physical development of the patient. Kinesiotherapy includes positional drainage, Klopf massage, vibration, special breathing exercises, active breathing cycles, forced expiratory technique, autogenous drainage, breathing with positive expiratory pressure. The last procedure is carried out under the supervision of a doctor, as complications including pneumothorax are possible. It is necessary to use bronchodilators, mucolytics (see below), and, if possible, amiloride (sodium blocker) and/or “pulmozyme” (DNAase produced by ).
| Patients with cystic fibrosis and their family members should be monitored in specialized centers. But in our country there are few such centers, and given the distances and the extreme difficulty of travel expenses, they become completely inaccessible to patients. Therefore, the principles of managing patients with CF should be familiar to doctors in a wide medical network. |
Pulmonary pathology. Frequent use of antibiotics. They should be prescribed at early signs of inflammation activation with courses lasting up to 2-3 weeks. Many antibiotics effective against pseudomonas require intravenous administration. For pseudomonas infection (according to the antibiogram!), aminoglycosides, third-generation cephalosporins, and fluoroquinolones are effective. Some of the cephalosporins and tobramycin can be inhaled. The latter at a dose of 300 mg, regardless of age, once a day. Cotrimaxazole is effective for staphylococcal contamination, but its clearance in patients with CF is increased, so the usual therapeutic dose must be increased.
Mucolytics are an indispensable attribute of CF therapy. Prescribed both orally and by inhalation: N-acetylcysteine 300-1200 mg/day. (An overdose of the drug leads to lysis of the mucous membrane.) Bronchoscopic administration of mucolytics followed by aspiration of secretions and antibiotics at the end of the bronchial lavage procedure (with ultrasound dispersion) is an effective way of endoscopic administration of drugs. Modern techniques of transnasal flexible bronchoscopy allow the procedure to be used on an outpatient basis.
In cases of bronchospastic syndrome - inhalation of beta-mimetics (long-term use is fraught with the development of arrhythmias and dilated cardiomyopathy), as well as corticosteroids (systemically or inhaled) in order to reduce inflammatory processes in the lungs (side effects - osteoporosis, excess weight, infectious complications), non-steroidal anti-inflammatory drugs. These drugs reduce the inflammatory reactions of the bronchial tree, which sometimes cause more harm than the infectious agent itself. From this point of view, the use of alpha-one antitrypsin, a serum leukocyte protease inhibitor, and anticytokines (primarily anti-interleukins IL2, IL8) is justified. Alternative chloride channels are opened by ADP and UDP (uridine diphosphate).
In North America and Europe, lung or heart-lung complex transplants are performed, and genetic engineering approaches are being developed to correct the function of the mutant gene by using pneumotropic viruses with genetic constructs built into them. In 1998, a gene therapy program for CF was launched in Russia.
Pancreatic insufficiency. Good nutritional status is one of the main goals in the treatment of children with CF. Therefore, continuous enzyme therapy is necessary. Patients with good physical development have a better prognosis. They are more active, tolerate physical activity better, and have better indicators of respiratory function and immunity. Pancreatin, mezim-forte, panzitrat, and Creon are effective (increasingly). The last two are in the form of granules and microtablets with a pH-sensitive shell. The dose is individual. Usually they start with 2–6 thousand units. lipase per kg weight/day. They increase gradually, based on the characteristics of the stool and the child’s weight. Exceeding the dose leads to irritation of the intestinal mucosa and inflammation. In extremely rare cases, post-inflammatory strictures of the colon are possible. A good effect in case of liver damage (cholestasis, precirrhosis, cirrhosis) is provided by the administration of ursodeoxycholic acid (ursosan) in combination with taurine, which promotes the excretion of bile acids, which facilitate the digestion of fats.
Principles of patient management, control studies. Nutrition should exceed age-specific caloric norms by 10–15%, and the introduction of multivitamins and microelements is mandatory. Protein diet without fat restriction, but with adequate replacement therapy with modern microspherical enzymes with a pH-sensitive shell.
Body weight control. Weight loss or a flat weight curve indicates poor enzyme supply or an exacerbation of a chronic bronchopulmonary process.
Bacteriological examination of sputum with an antibiogram or a throat smear once every 6 months and after an exacerbation of the bronchopulmonary process or when the color of the sputum changes (green, mixed with blood).
HbA1 - glycosylated hemoglobin. In children over 8 years old, determine 1-2 times a year. With reduced glucose tolerance - more often.
chest x -ray As a control - once a year.
Echocardiogram (especially the right side, pulmonary artery) at least once a year.
ECG. 1–2 times a year. According to indications - more often.
Functional tests of the lungs. External respiration function (usually from 6 years of age, the age of cooperative participation) and blood gases - once a month and after an exacerbation of the bronchopulmonary process.
Body plethysmography. From 8 years old - 1-2 times a year. According to indications - more often. The function of external respiration is not examined in the terminal stage, since in the pre-final period the tests are burdensome for the patient and do not affect the treatment.
liver cirrhosis is suspected , ultrasound examinations, liver function tests, prothrombin, and less commonly, biopsy.
- Complications and outcomes
Nasal polyps. Steroids inhaled or in the form of ointment applications. Surgical treatment is not advisable (the likelihood of relapse is high).
Pneumothorax. Develops in older children and adults. The likelihood of recurrence is high. Rest is required; if the volume is less than 10% of the lung volume, a minimum of manipulation is required. For tension pneumothorax - drainage, pleural puncture. In case of relapses - removal of the bullous lobe, pleurodesis.
Atelectasis. Bronchoscopy with bronchoalveolar lavage and administration of mucolytics, antibiotics, and breathing exercises are required.
Pneumonia. General principles of therapy. Drainage measures are extremely important.
Hemoptysis. As a symptom, it is overestimated and causes unjustified panic. It looks like an admixture of blood to sputum, most often caused by damage to the bronchial mucosa. For pulmonary hemorrhage (300 or more ml at one time or more than 100 ml in 3 days) - angiographic embolization or occlusion of the bleeding vessel. In case of failure, ligation of the vessel or resection of the segment (lobe).
Pulmonary heart. With adequate therapy, it develops as a manifest form only in adults. They are also characterized by heart rhythm disturbances. Digitalis is ineffective for “cor pulmonale”; it is advisable to use Corinfar, nifedipine and hydralazine (the latter is dangerous due to possible autoimmune reactions).
Aspergillosis. Associated with corticosteroid therapy. If aspergillus is found in sputum by chance and does not manifest itself clinically, then no treatment is required. Therapy is prescribed for widespread bronchiectasis, dilatation of the bronchi, increasing pulmonary symptoms, especially with signs of torpid obstruction, increased total IgE and specific IgE.
Allergies and asthma. In 25–48% of patients, a combination of CF and asthma is observed.
Salt deficiency exicosis. It can occur not only in newborns, but also in children of different ages and adults, especially in the hot season. Prevention - drink plenty of fluids and sufficient salt intake (3-8 g/day).
Diabetes mellitus is pancreatogenic. It develops very slowly, gradually. It is observed in 2% of children and 15% of adults with CF.
Bleeding from the stomach and from varicose veins of the esophagus (with cirrhosis of the liver) . Endoscopic sclerosis of varicose veins, partial resection of the spleen, and shunt operations are performed.
Gallbladder stones. Endoscopic removal is less risky than laparotomy, which has a higher chance of pulmonary complications.
The equivalent of meconium ileus in adolescents and adults is partial distal intestinal obstruction with thick, viscous stools. For ileus that does not require surgical intervention, wash with gastrografin, hypaque, N-acetylcysteine. If there is no effect, surgical intervention is required.
Pneumatosis of the intestinal wall can be detected accidentally and does not itself require intervention.
Rectal prolapse is very rare with adequate enzyme replacement therapy.
Pulmonary osteoarthropathy . Along with deformations of the terminal phalanges, periostotic pain may appear in the long tubular bones. To alleviate the condition, non-steroidal anti-inflammatory drugs are prescribed.
Chest deformities develop as a result of pulmonary pathology.
Thus, the combined efforts of pediatricians, therapists, bronchologists, gastroenterologists, nutritionists, psychologists, social workers, subject to early diagnosis and adequate therapy, interested participation in the treatment of the patient and his relatives, are already helping to change the quality of life and increase its duration in patients with CF.
Literature
1. Kapranov N. I., Rachinsky S. V. Cystic fibrosis. M., 1995, p. 188. 2. Kapranov N.I., Kashirskaya N.Yu. Current problems of cystic fibrosis. Pediatrics, 1998, No. 1, p. 61-66. 3. Consensus on cystic fibrosis. Medical newspaper, 1995, No. 41, 02.06.95. 4. Petrova N.V. Materials of the scientific-practical conference of the Russian Children's Clinical Hospital, M., 1995, p. 96. 5. Potapova O. Yu. Molecular genetic analysis of cystic fibrosis in Russia. Abstract of Ph.D., St. Petersburg, 1994, p. 24. 6. Becker D. Vademecum Labordiagnostic. Berlin, 1987, ss. 334. 7. Dean T., Dai Y., Shute K., ea Pediatric Research, 1993, Vol. 34, p. 159-161. 8. Dodge J, Brock D, Widdicombe J. Cystic Fibrosis. UK, 1994, p. 3. 9. Fanconi G., Uelinger E., Knauer C. Das Coeliakie-Syndrom bei angeborener zystischer Pancreasfibrose und Bronchiektasien. Wiener med. Wochenshrift, 1936, Bd. 86, ss. 753-756. 10. Fick R., Rabbins R. Pediatric Research, 1995, Vol. 20, p. 1258. 11. Hodson M., Geddes D. Cystic fibrosis. London, 1995, p. 439. 12. Illing St. Mukoviszidose. In the book. Paediatrie. Stuttgart, 1993, ss. 437-440. 13. Kopelman H., Davies M. Cystic fibrosis. in: Current Pediatric Therapy, Philadelphia, 1993, 4th Ed. pp. 135-139. 14. Leiber G. Die klinischen Syndrome. Muenchen, 1993, Bd. 1.2. 15. Welsh M., Smith A. Cystic fibrosis, NY, 1995, p. 36-43. 16. WHO/HGN/IGF(M)A. Guidelines for the Diagnosis and Management of CF., Geneve, 1996, p. 59.
Disease severity
In determining the severity of cystic fibrosis, signs of damage to the respiratory system play a major role.
Depending on their severity, the following degrees of severity of the pathology are distinguished:
- I - cystic fibrosis is characterized by minor changes of a non-permanent nature, cough and shortness of breath appear only during physical exertion;
- II - manifested by a chronic inflammatory process in the bronchi, constant moderate shortness of breath and deformation of the phalanges of the fingers. On auscultation, moist rales are heard;
- III - manifested by progressive damage to the bronchi and lungs, with the development of foci of fibrosis and sclerosis, the appearance of bronchiectasis, and the appearance of signs of cardiac and respiratory failure;
- IV - accompanied by severe heart and lung failure.
Assessing the symptoms, the doctor adjusts the dosage of drugs, thereby ensuring complete treatment of cystic fibrosis.
Forms and manifestations of the disease
In cystic fibrosis, all exocrine glandular formations are affected, regardless of their size. But the severity of pathological changes in different organs usually differs. Taking this into account, several clinical forms of the disease are distinguished:
- Meconium intestinal obstruction. It develops in infants in the first days of life and is caused by a pronounced thickening of their primary feces (meconium).
- Bronchopulmonary form associated with disruption of the glands of the bronchial epithelium and obstruction (clogging with mucus) of the final sections of the respiratory system. It manifests itself as recurrent bronchopneumonia with the formation of bronchiectasis and chronic obstructive pulmonary disease. (COPD).
- The intestinal form, which also includes a picture of damage to the pancreas (pancreatitis) with the development of enzyme deficiency.
- Biliary cirrhosis of the liver associated with obstruction of the biliary tract due to severe thickening of bile.
There are also very “mild” forms of the disease, when the existing disorders do not so severely affect the quality of life and are not considered potentially life-threatening conditions. For example, in men, cystic fibrosis can manifest as isolated obstructive azoospermia in the form of infertility due to obstruction of the vas deferens. There are abortive forms with damage to the sweat glands, sinusitis, chronic pancreatitis without a tendency to cyst formation, etc.
In clinical practice, the terminology of the International Classification of Diseases, 10th revision (ICD-10) is used. Cystic fibrosis has the code E84 and is divided into forms with pulmonary, intestinal and other manifestations. There is also a subsection “Unspecified cystic fibrosis”. At the same time, the doctor has to code only the most severe disorders, because in 70% of cases there is a mixed (pulmonary-intestinal) form of the disease.
When making a diagnosis, the severity of existing disorders is also indicated and complications that have already developed are described.
Diagnosis of cystic fibrosis
Timely diagnosis of cystic fibrosis is of great importance for a child’s life.
Before a diagnosis of cystic fibrosis is made, the following measures are taken:
- study of hereditary history, early symptoms and clinical manifestations of the disease; urine and blood analysis (general);
- coprogram is a study of feces for the content of starch, muscle fibers, fiber, fat, with the help of which the degree of disruption of enzymatic processes in the glands of the digestive tract is determined;
- sputum examination (microbiological);
- X-ray examination of the lungs - to identify sclerotic and infiltrative changes in their tissues; bronchography – detect bronchial defects;
- bronchoscopy - determine the presence of viscous and thick thread-like sputum in the bronchi;
- spirometry - determine the functional state of the lungs by measuring the speed and volume of exhaled air;
- molecular genetic testing - DNA or blood samples are analyzed to identify mutations in the cystic fibrosis gene;
- a sweat test is the main test for cystic fibrosis, with the help of which the patient is diagnosed with an increased content of sodium and chlorine ions in the sweat;
- prenatal diagnosis of cystic fibrosis - newborn children are examined for congenital and genetic diseases.
The intestinal form of cystic fibrosis must be distinguished from intestinal absorption disorders observed with disaccharidase deficiency, dysbacteriosis and enteropathy. The pulmonary form is differentiated from bronchial asthma, pneumonia of other origins, whooping cough and obstructive bronchitis.
Recovery forecasts
Scientists and pharmacists are constantly working to create new drugs that can normalize the concentration of chlorides in the body and improve lung function. The main goal is to restore CFTR function using corrective medications. At the moment, the hereditary disease cannot be completely cured, but with sufficient medical support it can be controlled. The later the signs of the disease appear, the better the prognosis for the patient. It is difficult to give an accurate prognosis for life expectancy with such a diagnosis. Much depends on the severity of the genotype and the type of its mutation, the rate of occurrence of complications. Treatment of cystic fibrosis in children remains a pressing issue. There are about 3,500 people in Russia with this diagnosis. The average lifespan for uncomplicated stages is 30 years.
Treatment of cystic fibrosis
How to treat cystic fibrosis? Treatment for cystic fibrosis depends on the form of the disease. If the intestinal form of the disease predominates, then a diet is indicated. Food should contain a large amount of proteins (eggs, cottage cheese, fish and meat). The consumption of fats and carbohydrates is sharply limited (only their easily digestible forms are allowed). It is necessary to exclude coarse fiber and milk from the diet (for lactose intolerance). You should also take vitamins and drink plenty of fluids (especially in summer).
Replacement therapy for this form of the disease includes taking enzyme preparations (Mezim, Festal, Panzinorm and a number of others). The frequency and dosage of medications is determined by the attending physician based on the severity of the disease and the severity of excretory insufficiency of pancreatic function. The effectiveness of the treatment is judged by the restoration of the patient’s weight, the absence of neutral fat in the stool, the disappearance of pain and the normalization of stool. Therapy for the pulmonary form of cystic fibrosis is aimed at restoring bronchial patency, reducing sputum viscosity and eliminating the inflammatory process. For this purpose, the prescription of mucolytics (acetylcysteine, mucosolvin), which are used in the form of inhalations or aerosols throughout the patient’s life, is indicated.
If inflammation of the lungs or bronchi develops, antibiotic therapy is indicated. Preference is given to broad-spectrum drugs that are active against most bacteria. Medicines are prescribed both in tablet form and by injection. The daily and course dose is determined by the attending physician based on the individual characteristics of the patient.
Steroidal anti-inflammatory drugs are used according to indications. They effectively reduce the activity of the inflammatory process, eliminate spasm of bronchial smooth muscles, relieve swelling, thereby increasing the lumen of the airways, normalizing breathing. In addition, physical therapy, vibration massage of the sternum and physical therapy are prescribed. For therapeutic purposes, bronchoscopic sanitation is performed using mucolytics. For acute pneumonia or bronchitis, antibacterial drugs are prescribed. In addition, the prescription of drugs that improve the nutrition of the heart muscle is indicated. Patients diagnosed with cystic fibrosis should be regularly examined by a physician and pulmonologist. Parents of a sick child should be trained in the rules of caring for him. The question of whether to prescribe preventive vaccinations for a child is decided on an individual basis. Children suffering from a mild form should receive sanatorium treatment for cystic fibrosis.
It is better for them not to go to kindergarten. As for school, the possibility of attending it depends on the general condition of the child.
Why does the disease develop?
Currently, there is no doubt that the only cause of cystic fibrosis is unfavorable heredity. This disease cannot occur under the influence of negative external factors or due to infection. The gene mutation that causes it is recessive, that is, it can be present in a latent form in any person and is detected only after a special genetic study. The disease occurs only if the father and mother turn out to be latent carriers of the damaged gene, and the child inherits it from both parents.
The disease is quite rare: in our country it is found in no more than three hundred people a year. Pathological changes occur in the pancreas, sweat and salivary glands, respiratory system, and digestive tract. Disturbances in the functioning of organs can appear already at the stage of intrauterine development. Patients diagnosed with cystic fibrosis must remain under medical supervision for the rest of their lives.
How to make an appointment with a doctor
Cystic fibrosis in children is accompanied by medical supervision. If you suspect hereditary factors, the presence of characteristic symptoms and the manifestation of signs, you should seek medical help. JSC "Medicine" (clinic of academician Roitberg) provides consulting services to specialists, conducts comprehensive health diagnostics and can offer qualified support for children with severe hereditary diagnoses. The clinic is located in the Central Administrative District and close to the Mayakovskaya metro station, as well as Novoslobodskaya, Chekhovskaya, Belorusskaya, and Tverskaya. Come for routine and preventive examinations at the address: 2nd Tverskoy-Yamskaya Lane, building 10. To make an appointment with a pediatrician, you need to call the number.
The clinic employs a staff of doctors of various specialties who help at all stages of treatment of the disease. A special atmosphere of trust creates a comfortable psychological microclimate for young patients. Cystic fibrosis in children has different clinical recommendations for correcting the general condition. A hereditary disease makes changes to the daily routine, constant therapy and procedures are required - these are the main provisions. At JSC “Medicine” (academician Roitberg’s clinic) they work with patients both in inpatient and outpatient settings.