pharmachologic effect
Somatotropic hormone.
Stimulates skeletal and somatic growth, and also has a pronounced effect on metabolic processes. Stimulates the growth of skeletal bones, affecting the epiphysis plates of tubular bones, bone metabolism in children. Helps normalize body structure by increasing muscle mass and reducing body fat. In patients with growth hormone deficiency and osteoporosis, replacement therapy leads to normalization of mineral composition and bone density. Increases the number and size of cells in muscles, liver, thymus, gonads, adrenal glands, and thyroid gland. Stimulates the transport of amino acids into the cell and the synthesis of proteins, reduces the concentration of cholesterol, affecting the profile of lipids and lipoproteins. Suppresses insulin release. Promotes the retention of sodium, potassium and phosphorus. Increases body weight, muscle activity and physical endurance.
Shereshevsky-Turner syndrome (STS) is a chromosomal disease based on complete or partial X-monosomy, present in all cells or in a mosaic version. The main clinical signs of STS include anomalies of physical development, short stature and sexual infantilism [1-3].
Growth impairment is observed in 95% of cases of STS and is manifested by reduced weight and height indicators at birth, low growth rates and the absence of a growth spurt in adolescence [4–6]. The average final height in STS varies in different populations from 140 to 147 cm [5, 7].
Short stature often becomes a serious psychological problem and a cause of social maladjustment [8]. The development of a genetic engineering method for producing recombinant growth hormone (r-GH) has made a real revolution in the treatment of children with various types of short stature. It became possible, in particular, to increase the growth rate in children with STS and thereby significantly improve the growth prognosis.
The purpose of this work was to evaluate the effectiveness and safety of a two-year treatment of girls with STS with the domestic genetically engineered growth hormone drug (somatotropin) Rastan.
Material and methods
29 girls with a karyotyping-confirmed diagnosis of TTS were under observation.
Chronological age at diagnosis was 7.9±1.2 years (0÷13.1 years), at the time of initiation of therapy - 9.8±1.6 years (5.0÷15.1 years). Bone age corresponded to 7.5±1.1 years (4÷11 years).
Patient height was measured using a mechanical Harpender stadiometer (Holstan Ltd, Crymych, Dyfed, UK) to the nearest 0.1 cm. Body weight was measured using an electronic floor scale. Growth indicators were assessed in SDS.
The SDS of height and growth velocity for children with STS was calculated twice: using percentile tables for healthy children [9] and for children with STS [10]. The stage of sexual development was determined according to the classification of J. Tanner [11]. The degree of differentiation of the bone skeleton was assessed according to the standards developed by W. Greulich, S. Pule [12].
All examined patients received r-GH Rastan (Pharmstandard-UfaVITA OJSC) as a growth-stimulating therapy in the form of a lyophilisate of 1.33 mg to prepare a solution for subcutaneous administration. The strain and technology for producing somatotropin were developed at the Institute of Bioorganic Chemistry of the Russian Academy of Sciences under the leadership of Academician of the Russian Academy of Sciences A.I. Miroshnikova. r-GH was prescribed at a dose of 0.05 mg/kg body weight daily subcutaneously in the evening (20.00-22.00 hours). The duration of treatment was 24 months.
Growth dynamics, SDS growth, annual growth rate and SDS growth rate during the therapy period were used as criteria for assessing the effectiveness of the drug.
Statistical processing of the obtained data was carried out on a personal computer using Statistica for Windows, version 5.5 and Microsoft Excel (2002). Data are presented as mean ± standard deviation (min÷max). Differences were considered statistically significant at p<0.05.
results
Impaired growth and physical development were identified in all patients with STS. The mean height SDS compared to the healthy population was –3.0±1.1, and the mean height SDS calculated for patients with STS was –0.99±0.8. The characteristics of patients with STS are presented in Table. 1.
The average SDS value of parental height was 0.3±0.1 and did not differ from that in the healthy population.
Among the concomitant diseases, 9 girls had scoliosis, 12 had pathology of the cardiovascular system, and 8 had kidney pathology. However, the severity of these conditions was unlikely to significantly influence growth.
The average level of fasting blood plasma glucose was 4.5±0.6 mmol/l, the average level of glycated hemoglobin HbA1c was 5.4%, which was not outside the normal range.
The first Tanner stage of puberty was registered in 27 girls, and the second stage in 2 patients. When assessing the condition and function of the thyroid gland, autoimmune thyroiditis with a decrease in thyroid function was detected in 2 girls, for which therapy with levothyroxine sodium was prescribed, the dose of which varied from 50 to 100 mcg and was adjusted depending on the concentration of TSH and free thyroxine in the blood serum. Before starting r-GH therapy, only one girl received replacement therapy with microfollin (at a dose of 1/8 tablet per day). Another girl showed spontaneous sexual development (Tanner stage of mammary gland development B2) at the age of 12 years after 6 months of treatment with r-GH. The results of treatment with r-GH in patients with STS are presented in Table. 2.
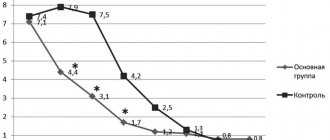
Before the start of r-GH treatment, the height of girls with STS increased by an average of 4.2±0.6 cm/year. The average growth rate in the first year of treatment was 8.7±0.6 cm/year, in the second year of treatment - 6.2±1.2 cm/year. Over 2 years of r-GH therapy, girls gained an average of 14.9 cm in height.
The increase in height SDS was significant after 12 months (p=0.002) and 24 months (p=0.003) of r-GH treatment. In just 2 years, girls' height increased on average by 0.84 SD.
SDS growth rates also significantly increased after 12 months (p=0.001) and 24 months (p=0.004) of r-GH treatment.
Over 2 years of therapy, glycemic levels did not go beyond the normal range; the average HbA1c level in patients with STS after 2 years of treatment with r-GH was 5.8%.
Discussion
Short stature is a characteristic feature of TTS and is formed as a result of reduced growth rates in childhood and adolescence [7]. The results of numerous studies [4, 6] conducted in many countries indicate that girls with TTS who did not receive any growth-promoting therapy have a final body height that is 20 cm lower than the population average for girls.
In recent decades, it has been proven that the leading role in the pathogenesis of short stature in STS is played by direct genetic disorders caused by deletion of the SHOX gene located on the X chromosome [13], leading to disruption of the spontaneous secretion of growth hormone (GH), insensitivity to GH and disturbances in the system GH/insulin-like growth factor type 1.
The growth retardation of the 29 girls with STS we examined was significant. However, when compared with the population of girls with STS, it turned out that the growth retardation in these girls is slightly greater than the average typical for the standards of this syndrome (SDS 0.99 ± 0.37).
Currently, the most effective treatment for short stature in patients with STS are r-GH drugs prescribed for a long period. In many countries, research programs are exploring the potential of r-GH (alone or in combination with sex hormones) for the treatment of patients with TS with the aim of accelerating growth in childhood and increasing final height [14–16].
Treatment with r-GH in childhood in girls with STS is not replacement, but may have a positive effect [17].
It is generally accepted to treat patients with STS with r-GH at a dose of 0.375 mg/kg/week or 0.05 mg/kg/day, which is 50% higher than the standard dose used in children with somatotropic insufficiency.
Our treatment of girls with STS with r-GH at a dose of 0.05 mg/kg/day undoubtedly gave positive results. If before treatment girls grew on average about 4 cm per year, then during therapy in the 1st year the patients gained an average of 8.7 cm (+0.5 SD), in the second year 6.0 cm (+0 .34 SD).
Other studies that examined the dependence of the growth effect on the dose of r-GH confirmed the greater effectiveness of high doses. There are reports of a significant increase in height (10.6 cm) in 12 girls with STS who were treated with r-GH using a stepwise dose-increasing regimen. This regimen involved the use of r-GH at an initial dose of 0.7 units/kg/week, increasing to 1.4 units/kg/week, and then to 2.1 units/kg/week every 6 months, which made it possible to constantly maintain growth rate, 2 times higher than that before the start of therapy. At the same time, the increase in height in 17 girls with TTS who received a fixed standard dose of 0.9 units/kg/week was only 5.2 cm.
According to the results of one of the long-term studies (Dutch study), in which r-GH in doses of 0.045, 0.067 and 0.089 mg/kg/day was administered daily for 7 years, it was shown that the highest doses and the longest treatment led to an increase in final height from 159.1, 161.8 and 167.2 cm, respectively, and the maximum doses of r-GH provided the most pronounced increase in height (16 cm versus 12.5 cm) [18].
The final height gain, defined as the difference between the final height and that predicted before the start of therapy, after treatment with doses of r-GH (0.05 mg/kg/day) varied among different authors from 3.1 to 21.4 cm [15, 19— 27]. It should be noted that an important characteristic feature of all these studies was significant individual fluctuations in the final growth achieved. In our study, girls gained an average of 14.7 cm in height over 2 years of r-GH therapy, which corresponds to 0.84 SD, but no one has yet achieved final height. In earlier studies conducted at the ERC (N.N. Volevodz, 2005), the final height of patients with STS who received r-GH treatment did not reach a socially acceptable level, and the height of 3 patients who completed treatment was only 139.1. 140.5 and 144.9 cm [1]. This circumstance was associated with late administration of r-GH (on average 12 years) and a short period of treatment (on average 12 months), which did not allow them to achieve socially acceptable height.
Similar data were obtained from the National Cooperative Growth Study (NCGS). It states that the peak age for r-GH prescription is between 10 and 12 years of age. Thus, treatment started belatedly does not allow, unfortunately, r-GH to fully demonstrate its therapeutic effect and help the majority of these children achieve a height of 150 cm [17].
Currently, there are results of the use of r-GH in young children with STS. M. Davenport et al. [28] observed 88 girls with STS aged from 9 months to 4 years, of whom 45 received r-GH treatment at a dose of 0.05 mg/kg/day for 2 years, and 43 were in the control group and did not receive therapy . The children's height at the start of treatment was –1.4 and –1.8 SDS, respectively. By the end of the 2nd year of observation, growth rates in the treatment group corresponded to –0.3 SDS (+1.1 SDS), and in the control group –2.2 SDS (–0.5 SDS). Consequently, girls who did not receive r-GH treatment had worse growth indicators after 2 years, while the height deficit in girls with treatment decreased.
Thus, the results of numerous studies [29, 30] and multiple regression analyzes have shown that growth rate in the 1st year of therapy is considered an important prognostic indicator of the effectiveness of treatment in girls with STS. In addition, factors that positively influence the results of r-GH therapy include the dose and frequency of injections, the time of pubertal induction and the dose of estrogens used. The dependence of the degree of increase in final height on the duration of treatment was revealed.
When assessing the safety of therapy with r-GH Rastan, we did not note a single case of serious adverse events that would lead to discontinuation of the drug, as well as unexpected ones not typical for somatotropin. Currently, many publications raise concerns that treatment with r-GH may increase the risk of developing a number of complications inherent in STS, such as impaired carbohydrate tolerance, osteoarticular changes, dyslipidemia and cardiovascular diseases. Data from the KIGS study confirm that STS has a higher incidence of side effects than idiopathic GH deficiency (148 per 1000 years of treatment compared with 89/1000 years of treatment). In STS, diabetes mellitus types 1 and 2, scoliosis and epiphysiolysis of the femur were more often detected, and in terms of the frequency of headaches and intracranial hypertension, patients with STS were second only to patients with craniopharyngioma. However, all these complications themselves are characteristic of STS, and can partly be considered as concomitant diseases.
conclusions
1. Treatment of girls with TTS with the genetically engineered somatotropin drug Rastan allows one to achieve an increase in growth rates. During the 1st year of treatment, the increase in height was 8.7 cm, in the 2nd year - 6.0 cm (the average increase over 2 years of treatment was 14.7 cm).
2. During the study period of use of the drug r-GH Rastan, no adverse events were registered, which indicates its safety in childhood.
Directions for use and doses
Rastan is administered subcutaneously, slowly, once a day, usually at night. Injection sites should be changed to prevent the development of lipoatrophy.
It is recommended to dissolve the contents of the bottle in 1 ml of the supplied solvent, based on the calculated dose. To do this, remove the solvent with a syringe and inject it into the bottle with the drug through the stopper. Gently rock until the contents of the bottle are completely dissolved. Sharp shaking is unacceptable. The prepared solution is stored in the bottle for no more than two weeks at a temperature from 2 ºС to 8 ºС.
Doses are selected individually, taking into account the severity of growth hormone deficiency, weight or body surface area, and effectiveness during therapy.
In children with insufficient secretion of growth hormone, a dose of 25-35 mcg/kg/day is recommended. (0.07-0.1 IU/kg/day), which corresponds to 0.7-1 mg/m2/day (2-3 IU/m2/day). Treatment begins as early as possible and continues until puberty and/or until bone growth plates close. It is possible to stop treatment when the desired result is achieved.
For Shereshevsky-Turner syndrome, for chronic renal failure in children, accompanied by growth retardation, a dose of 45-50 mcg/kg/day is recommended. (0.14 IU/kg), which corresponds to 1.4 mg/m2/day (4.3 IU/m2/day). If growth dynamics are insufficient, dose adjustment may be required.
For growth hormone deficiency in adults, the initial dose is 0.15-0.3 mg/day. (which corresponds to 0.45-0.9 IU/day) with its subsequent increase, depending on the effect. When titrating the dose, the level of insulin-like growth factor (IGF-I) in the blood serum can be used as a control indicator. The maintenance dose is selected individually, but usually does not exceed 1 mg/day, which corresponds to 3 IU/day. Lower doses are recommended for the elderly.
Issues of effectiveness and safety of the use of growth hormone drugs in pediatric practice
The use of growth hormone (GH) drugs in therapeutic algorithms for diseases accompanied by disturbances in the dynamics of growth indicators is today due to their wide availability despite their high cost. Recombinant human GH, obtained biosynthetically, has been used in pediatric practice as a means of stimulating growth and as a metabolic regulator of impaired metabolic processes since 1985. At the same time, somatropin preparations (INN growth hormone preparations) have certain indications with proven effectiveness. The main indication for the use of somatropin is hypopituitarism. It is the treatment of GH deficiency in hypopituitarism with somatropin preparations that is pathogenetically justified and carried out for replacement purposes.
The use of GH in other types of short stature is possible, but prognostically unpredictable. To date, there is data on the effectiveness of GH treatment for children with intrauterine growth retardation, familial short stature, Shereshevsky–Turner, Prader–Willi, Russell–Silver syndromes, Fanconi anemia, glycogenosis, chronic renal failure, skeletal dysplasia, and cystic fibrosis [1].
Since 2003, biosynthetic GH has been used in the United States to treat children with idiopathic short stature. Age at initiation of GH therapy and response to GH therapy in the first year of treatment are major determinants of final height in idiopathic short stature. It has been suggested that children with a good growth response in the first year of GH treatment for idiopathic short stature are likely to have good final height using even low doses of GH [2]. However, observational results showed that the individual effect of therapy remains difficult to predict. The large variability of auxiological data, age, growth and bone age at the start of therapy, and growth rate in the first year of treatment determine multivariate analysis, which is difficult to predict.
It is quite difficult to predict the response to GH treatment even with replacement therapy. Evaluation of the effectiveness of GH treatment for somatotropic insufficiency using auxiological parameters showed that the growth response is variable and different in each specific case [3]. A characteristic feature of the treatment of somatotropic deficiency is that, according to observations of patient therapy, the result of the first year of treatment is higher than in subsequent years. It was noted that the only clinically significant predictor of growth response to the start of treatment in the first year is the age at the start of therapy. While neither the GH peak during stimulation tests, nor gender, nor height at the start of therapy, body mass index (BMI), bone age, or GH dose affect the growth rate in the first year of therapy. Earlier diagnosis and treatment of GH allows for better final growth in severe cases of GH deficiency. It was shown that the best response to treatment was observed in children with severe GH deficiency [4]. But, according to other data, final height does not differ significantly between patients with severe GH deficiency and patients with partial GH deficiency [5].
A number of studies have shown that parental height is one of the indicators that predicts final height in patients with GH deficiency receiving GH replacement therapy. It has been noted that GH treatment has better efficacy in patients whose families do not have relatives with short stature [6].
Although treatment with GH drugs is not always associated with the achievement of genetically determined height, the increase in height in the first year of treatment at the age before the onset of puberty correlates with the overall increase in height during therapy. This confirms the importance of starting treatment before puberty [7].
Currently, a number of studies are being conducted to select the necessary combination of drugs, the combination with which GH will improve treatment results. Thus, it has been shown that the simultaneous administration of GH replacement therapy and a GnRH agonist for congenital GH deficiency in order to inhibit the initiation of sexual development has no advantages over the administration of GH alone [8].
The growth effects of GH in somatotropic deficiency in patients with isolated GH deficiency and in patients with multiple pituitary hormone deficiency are expressed to the same extent: 89% of patients with isolated GH deficiency and 81% of patients with multiple pituitary hormone deficiency achieve the predicted growth. Moreover, a greater increase in height is observed at the age before the onset of puberty [9].
Many years of experience in treatment with GH drugs have shown that treatment of children with short stature using a dose of somatropin calculated for the child’s body weight is accompanied by large variability in the growth response to GH therapy. The question of why children with GH deficiency, whose therapy is aimed at simply replacing the missing GH, have such different clinical outcomes, has not yet been resolved.
Research is underway aimed at searching for pharmacogenomic markers with prognostic significance of cell sensitivity to GH.
The change in the level of insulin-like growth factor 1 (IGF-1) after 1 month of treatment with GH in children with GH deficiency was studied and it was proven that there is a relationship between the polymorphism of the cellular regulator CDK4 and the degree of change in the concentration of IGF-1. Further study of the relationship between genomic markers and early changes in IGF-1 levels may allow the development of a strategy for rapid individual selection of GH dosage for congenital somatotropic deficiency [10].
Also, the final height of patients receiving GH treatment is influenced by the presence or absence of the SOCS2 polymorphism (rs3782415). Polymorphisms detected in the GHR, IGFBP3 and SOCS2 loci affect the growth outcomes of patients with congenital somatotropic deficiency receiving GH. The use of these genetic markers can identify patients who are genetically predisposed to less effective treatment [11].
The end result of such studies should be recognition of the importance of individualized GH dosing for each patient based on specific individual genomic characteristics. This will significantly improve therapy, which for many years has been based on the principle of “one dosage fits all.”
How much the therapeutic effectiveness of somatropin compares with its safety is a question that requires in-depth detailed analysis, the solution of which is possible by accumulating experience in the use of GH in the treatment of various forms of short stature.
Safety during treatment and adverse effects of GH treatment have been carefully monitored and reported in children with GH deficiency (both isolated and multiple pituitary hormone deficiencies) and in children with idiopathic short stature [12]. Based on available information, derived primarily from post-marketing studies supported by manufacturers of GH drugs, there is a low incidence (less than 3% of treated children) of side effects and an increasing favorable safety profile of GH. However, the full range of potential side effects of GH is not accurately diagnosed through post-marketing studies. This is due to the rather long duration of treatment, the changing characteristics of the patient and the inability to track adverse events after the end of the patient’s treatment [13–15].
A rare adverse event associated with GH treatment is intracranial hypertension. A higher risk of its development was noted in groups of patients with chronic renal failure, Shereshevsky–Turner syndrome and with organic causes of GH deficiency. Intracranial hypertension usually develops during the initial period of treatment or with an increase in the dose of somatropin drugs, and ceases with the end of GH therapy. Indications for fundoscopy by an ophthalmologist include symptoms suggestive of intracranial hypertension, such as severe headache, double/blurred vision, and vomiting. Treatment can often be restarted at lower doses of GH without return of symptoms.
Complications of somatropin therapy include changes in the skeletal system - epiphysiolysis and scoliosis. Epiphysiolysis is diagnosed with a frequency of 73 per 100,000 years of treatment and occurs less frequently in patients with isolated GH deficiency and idiopathic short stature compared to those patients in whom GH deficiency is observed due to intracranial neoplasms, craniopharyngeoma [16]. The average duration from the start of GH therapy to the onset of epiphysiolysis ranges from 0.4 to 2.5 years. It is recommended to regularly monitor the appearance of relevant symptoms, such as hip pain and/or knee pain, changes in gait, and in case of a positive result, a thorough examination and consultation with an orthopedist. Epiphysiolysis requires surgical intervention on the epiphysis of the femur.
Scoliosis progresses during GH treatment due to rapid growth and is not a direct side effect of GH. Scoliosis most often develops in the treatment of syndromic forms of short stature (for example, Shereshevsky–Turner syndrome and Prader–Willi syndrome) [17]. Progression of scoliosis was noted in 0.2% of children with idiopathic short stature or with isolated GH deficiency treated with GH [18]. Regular screening for the presence or progression of scoliosis is recommended for all patients receiving GH medications.
Data from scientific publications and our own practical clinical experience indicate the need to monitor glucose metabolism in patients receiving GH. This is due to the proven fact of insulin resistance during treatment with somatropin drugs. The incidence of type 1 diabetes mellitus (DM) does not increase with GH therapy. At the same time, it is known that patients with Shereshevsky–Turner and Prader–Willi syndrome have a high risk of developing non-immune forms of diabetes mellitus. A decrease in insulin sensitivity and a compensatory increase in insulin secretion with euglycemia is accompanied by impaired glucose tolerance and requires correction of metabolic disorders, and in some cases, discontinuation of GH drugs [19]. Monitoring for the potential development of diabetes mellitus with blood glucose and/or HbA1c testing should be included in the surveillance algorithm for all patients receiving GH drugs.
Pathophysiological and epidemiological observations raise concern that GH may increase the risk of developing malignancy during or after therapy. It is known that the implementation of the growth effect of GH is mediated by somatomedins, one of which is IGF-1. IGF-1 and GH are substances with mitogenic and anti-apoptotic activities, and their receptors are found in tumors. States of impaired and excessive GH secretion/action are associated with decreased and increased risk of malignancy, respectively. Reduction of IGF-1 through calorie restriction induces apoptosis and prevents or slows tumor growth [20–22]. Some limited epidemiological studies have correlated increased GH and IGF-1 levels with colon, breast, thyroid and prostate cancers in adults [23]. In general, literature data indicate a permissive/facultative, rather than causative, role of GR in tumorigenesis.
The incidence of new leukemia diagnosed during treatment or malignancies in general after treatment in children without associated risk factors does not increase compared with that in the general population. Despite data on the absence of the effect of GH on the appearance of a second neoplasm in children with a history of neoplasia [24, 25], the clause about the increased risk of developing a second neoplasm in patients receiving GH is currently included in the instructions for use of all GH preparations in USA. There are no data regarding the effects of GH on the risk of developing neoplasia in patients with diseases that are themselves considered to be at increased risk for neoplasms. The risk of neoplasms in patients receiving GH treatment has been reviewed by the Drugs and Therapeutics Committee of the Society of Pediatric Endocrinologists, and a key recommendation is that continued monitoring of all patients treated with GH is of paramount importance [26].
Before initiating growth hormone therapy in a child, parents should be informed of the uncertainty regarding long-term safety (adverse effects following therapy in adulthood).
A long-term study (average 17 years) of 6928 children with isolated growth deficiency, idiopathic short stature, or low gestational age who started GH treatment between 1985 and 1996 in France showed a 30% increase in all-cause mortality compared with population as a whole [27]. All cancer-related deaths were not increased, but standardized mortality rates were increased in the groups of patients with skeletal tumors, patients with circulatory disorders, and patients with cerebral hemorrhage. The use of GH doses greater than 50 mcg/kg/day is not recommended. Data from the same database in a recent study showed a significantly higher risk of stroke (especially hemorrhagic stroke) among patients treated with GH during childhood [28]. In contrast, a follow-up study of 2543 patients with isolated growth deficiency, idiopathic short stature or low gestational age from other European countries found no effect of GH exposure and/or dose on mortality or incidence of cardiovascular events [29].
Thus, the available data regarding the safety of GH therapy for children with various forms of short stature determine the need to develop a personalized algorithm for clinical follow-up of the patient and should include informing the child’s parents about the currently available information about adverse events both during treatment and in long-term periods of life.
The use of high doses of growth hormone drugs for syndromic forms of short stature should have a balanced approach to comparing the effectiveness and safety of treatment. Increasing the dose of GH increases the chances of long-term metabolic or malignant risks not found in studies to date. Changing patient characteristics, ethnicity, and rising rates of childhood obesity may increase the risk of developing type 2 diabetes in those receiving GH.
Certain side effects of GH drugs associated with accelerated growth (progression of scoliosis and epiphysiolysis) and other unknown mechanisms (intracranial hypertension) are rare, but require proactive explanation and careful monitoring.
In addition, one should be aware of the long-term consequences of hormonal treatment. Because studies of GH-naïve populations suggest that high normal levels of free IGF-1 (often found in children treated with GH) may increase cancer risks, potential associations between GH exposure and future risks of neoplasia require ongoing monitoring. Finally, the appropriate level of acceptable risk for the newest and potentially largest group of patients treated with GH—apparently healthy but short children—has yet to be determined [30].
Literatures
- Peterkova V. A. Pituitary dwarfism: diagnosis and treatment // Pediatrics. 2009. No. 87 (02). pp. 104–110.
- Ranke MB, Lindberg A, Price DA et al. KIGS International Board. Age at growth hormone therapy start and first-year responsiveness to growth hormone are major determinants of height outcome in idiopathic short stature // Horm Res. 2007. No. 68 (2). R. 53–62.
- Kelnar CJ Growth hormone for short children – whom should we be treating and why? // JR Coll Physicians Edinb. 2012. No. 42 (1). R. 32–33.
- Ranke MB, Lindberg A. KIGS International Board. Observed and predicted growth responses in prepubertal children with growth disorders: guidance of growth hormone treatment by empirical variables // J Clin Endocrinol Metab. 2010. No. 95 (3). R. 1229–1237.
- Cardoso DF, Martinelli CE Jr, Campos VC et al. Comparison between the growth response to growth hormone (GH) therapy in children with partial GH insensitivity or mild GH deficiency // Arq Bras Endocrinol Metabol. 2014. No. 58 (1). R. 23–29.
- Hilczer M., Smyczynska J., Lewinski A. Parentally-adjusted deficit of height as a prognostic factor of the effectiveness of growth hormone (GH) therapy in children with GH deficiency // Neuro Endocrinol Lett. 2006. No. 27 (1–2). R. 149–152.
- Reiter EO, Price DA, Wilton P. et al. Effect of growth hormone (GH) treatment on the near-final height of 1258 patients with idiopathic GH deficiency: analysis of a large international database // J Clin Endocrinol Metab. 2006. No. 91 (6). R. 2047–2054.
- Colmenares A., González L., Gunczler P., Lanes R. Is the growth outcome of children with idiopathic short stature and isolated growth hormone deficiency following treatment with growth hormone and a luteinizing hormone-releasing hormone agonist superior to that obtained by GH alone ? // J Pediatr Endocrinol Metab. 2012. No. 25 (7–8). R. 651–657.
- Darendeliler F., Lindberg A., Wilton P. Response to growth hormone treatment in isolated growth hormone deficiency versus multiple pituitary hormone deficiency // Horm Res Paediatr. 2011. No. 76. Suppl 1. R. 42–46.
- Stevens A., Clayton P., Tatò L. et al. Pharmacogenomics of insulin-like growth factor-I generation during GH treatment in children with GH deficiency or Turner syndrome // Pharmacogenomics J. 2014. No. 14 (1). R. 54–62.
- Braz AF, Costalonga EF, Trarbach EB Genetic predictors of long-term response to growth hormone (GH) therapy in children with GH deficiency and Turner syndrome: the influence of a SOCS2 polymorphism // J Clin Endocrinol Metab. 2014. No. 99 (9). R. 1808–1813.
- Wilson TA, Rose SR, Cohen P. et al. Update of guidelines for the use of growth hormone in children: The Lawson Wilkins Pediatric Endocrinology Society Drug and Therapeutics Committee // J Pediatr. 2003. No. 143. R. 415–421.
- Bell J, Parker KL, Swinford RD et al. Long-term safety of recombinant human growth hormone in children // J Clin Endocrinol Metab. 2010. No. 95. R. 167–177.
- Darendeliler F., Karagiannis G., Wilton P. Headache, idiopathic intracranial hypertension and slipped capital femoral epiphysis during growth hormone treatment: a safety update from the KIGS database // Horm Res. 2007. No. 68. Suppl 5. P. 41–47.
- Grimberg A., DiVall S., Polychronakos C. et al. Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency // Horm Res Paediatr. 2021. No. 86. R. 361–397.
- Mostoufi-Moab S., Isaacoff E. J., Spiegel D. et al. Childhood cancer survivors exposed to total body irradiation are at significant risk for slipped capital femoral epiphysis during recombinant growth hormone therapy // Pediatr Blood Cancer. 2013. No. 60. R. 1766–1771.
- Kim JY, Rosenfeld SR, Keyak JH Increased prevalence of scoliosis in Turner syndrome // J Pediatr Orthop. 2001. No. 21. R. 765–766.
- Cohen P., Bright GM, Rogol AD et al. Effects of dose and gender on the growth and growth factor response to GH in GH-deficient children: implications for efficacy and safety // J Clin Endocrinol Metab. 2002. No. 87. R. 90–98.
- Bareille P., Azcona C., Matthews DR et al. Lipid profile, glucose tolerance and insulin sensitivity after more than four years of growth hormone therapy in non-growth hormone deficient adolescents // Clin Endocrinol (Oxf). 1999. No. 51. R. 347–353.
- Aguiar-Oliveira M., Oliveira FT, Pereira RM et al. Longevity in untreated congenital growth hormone deficiency due to a homozygous mutation in the GHRH receptor gene // J Clin Endocrinol Metab. 2010. No. 95. R. 714–721.
- Orme SM, McNally RJQ, Cartwright RA, Belchetz PE For the United Kingdom Acromegaly Study Group: Mortality and cancer incidence in acromegaly: a retrospective cohort study // J Clin Endocrinol Metab. 1998. No. 83. R. 2730–2734.
- Speakman JR, Mitchell SE Caloric restriction // Mol Aspects Med. 2011. No. 32. R. 159–221.
- Cohen P., Clemmons DR, Rosenfeld RG Does the GH-IGF axis play a role in cancer pathogenesis? // Growth Horm IGF Res. 2000. No. 10. R. 297–305.
- Wilton P., Mattsson AF, Darendeliler F. Growth hormone treatment in children is not associated with an increase in the incidence of cancer: experience from KIGS (Pfizer International Growth Database) // J Pediatr. 2010. No. 157. R. 265–270.
- Mackenzie S., Craven T., Gattamaneni HR et al. Longterm safety of growth hormone replacement after CNS irradiation // J Clin Endocrinol Metab. 2011. No. 96. R. 2756–2761.
- Raman S., Grimberg A., Waguespack SG et al. Risk of neoplasia in pediatric patients receiving growth hormone therapy — a report from the Pediatric Endocrine Society Drug and Therapeutics Committee // J Clin Endocrinol Metab. 2015. No. 100. R. 2192–2203.
- Carel JC, Ecosse E, Landier F et al. Long-term mortality after recombinant growth hormone treatment for isolated growth hormone deficiency or childhood short stature: preliminary report of the French SAGhE study // J Clin Endocrinol Metab. 2012. No. 97. R. 416–425.
- Poidvin A., Touze E., Ecosse E. et al. Growth hormone treatment for childhood short stature and risk of stroke in early adulthood // Neurology. 2014. No. 83. R. 780–786.
- Savendahl L., Maes M., Albertsson-Wikland K. et al. Long-term mortality and causes of death in isolated GHD, ISS, and SGA patients treated with recombinant growth hormone during childhood in Belgium, The Netherlands, and Sweden: preliminary report of 3 countries participating in the EU SAGhE study // J Clin Endocrinol Metab . 2012. No. 97. R. 213–217.
- Allen DB Growth hormone therapy for short stature: is the benefit worth the burden? // Pediatrics. 2006. No. 118. R. 343–348.
E. B. Bashnina, Doctor of Medical Sciences, Professor O. S. Berseneva1
FSBEI HE Northwestern State Medical University named after. I. I. Mechnikova Ministry of Health of the Russian Federation, St. Petersburg
1 Contact information
special instructions
During treatment with Rastan, it may be necessary to adjust the doses of hypoglycemic drugs in patients with diabetes, manifestation of latent hypothyroidism may occur, and in patients receiving sodium levothyroxine, signs of hyperthyroidism may appear. During treatment, it is necessary to monitor the condition of the fundus, especially with symptoms of intracranial hypertension. Swelling of the optic nerve requires discontinuation of the drug. Detection of lameness during somatropin therapy requires careful monitoring. It is necessary to change the sites of subcutaneous injections due to the possibility of developing lipoatrophy. During kidney transplantation, treatment with the drug should be discontinued.
Provided there are no symptoms of increased intracranial pressure (headache, nausea, vomiting, blurred vision), Rastan does not affect the ability to drive vehicles or engage in other potentially hazardous activities that require increased concentration and speed of psychomotor reactions.
Rastan®
Treatment with somatropin should be carried out by physicians experienced in the diagnosis and treatment of patients with GH deficiency or Shereshevsky-Turner syndrome.
The maximum recommended daily dose should not be exceeded (see section "Dosage and Administration").
Stimulation of longitudinal growth can be carried out in children before the closure of the epiphyseal growth plates.
Growth hormone deficiency in adults persists throughout life and requires appropriate treatment, but there are currently no results from long-term therapy in adults.
Shereshevsky-Turner syndrome
In patients with Shereshevsky-Turner syndrome, during treatment with somatropin, it is recommended to monitor the proportional growth of the upper and lower extremities, and if increased growth is detected, the dose of the drug should be reduced to the lower limit of the dose range.
Girls with Shereshevsky-Turner syndrome usually have an increased risk of developing otitis media, and therefore should be monitored by an otolaryngologist.
Chronic renal failure Growth impairment in children with chronic renal failure should be accurately established before starting treatment with somatropin by monitoring growth against the background of optimal treatment for chronic renal failure for one year. During somatropin therapy, conservative treatment of chronic renal failure with traditional medications and, if necessary, dialysis should be continued. Somatropin therapy should be discontinued during kidney transplantation.
Pouder-Willi syndrome There have been reports of deaths in children with PWS with GH deficiency who were treated with somatropin and had at least one of the following risk factors: severe obesity, a history of respiratory failure, sleep apnea, or an unidentified respiratory tract infection.
A possible risk factor may be the male gender of the patient.
Patients with PWS with one or more of these factors are at high risk when using somatropin.
Before prescribing somatropin to patients with GH deficiency in combination with PWV, the potential risk-benefit ratio should be considered.
In patients with PWS, treatment with somatropin must necessarily be associated with a calorie-restricted diet. Patients with PWS should actively monitor their body weight both before and during somatropin use.
Tumors
Patients with a history of malignant neoplasms should be carefully examined for recurrence. If malignancy occurs or recurs, somatropin therapy should be discontinued.
Patients with GH deficiency secondary to the presence of brain tumors should undergo more frequent examinations to exclude progression and relapse of the underlying disease.
Somatropin should not be prescribed if any signs of active tumor growth are detected. Before prescribing somatropin, the tumor process must be in an inactive phase and antitumor therapy must be completed. If signs of tumor regrowth appear, administration of the drug should be stopped.
The development of secondary benign and malignant neoplasms has been reported in patients with childhood cancer and receiving somatropin therapy. The most common complication was the development of intracranial tumors, in particular meningioma, in patients who had previously received radiotherapy to the head for primary tumors. However, recurrence of primary tumors has not been reported in this category of patients.
Leukemia
Leukemia has been reported in children treated with somatropin. The relationship between the occurrence of leukemia and somatropin therapy has not been established.
Benign intracranial hypertension
If severe or recurrent headaches, blurred vision, nausea and/or vomiting occur, fundoscopy is recommended to identify possible papilledema. When the diagnosis is confirmed, the presence of benign intracranial hypertension should be assessed and, if the diagnosis is confirmed, somatropin therapy should be discontinued.
To date, there are no clear guidelines for the use of growth hormone in patients with corrected intracranial hypertension. However, clinical experience suggests that resumption of somatropin treatment in many cases does not lead to relapse of intracranial hypertension. If the use of somatropin is resumed, careful monitoring is necessary for the possible appearance of symptoms of intracranial hypertension.
Epiphysiolysis
In patients with endocrine disorders, including GH deficiency, epiphysiolysis of the heads of long bones may be more common. A thorough examination is necessary if the child develops lameness during treatment.
Hypopituitarism
Patients with hypopituitarism (deficiency of several pituitary hormones) in the case of standard hormone replacement therapy with the introduction of somatropin should be under strict supervision.
Thyroid function
When treated with somatropin, increased conversion of thyroxine (T4) to triiodothyronine (T3) was detected, which may cause a decrease in the concentration of T4 and an increase in the concentration of T3 in the blood plasma.
In healthy volunteers, as a rule, the concentrations of thyroid hormones in the blood plasma remained within normal limits. The effect of somatropin on thyroid hormone concentrations may be of clinical significance in patients with central subclinical hypothyroidism who may potentially develop hypothyroidism. On the other hand, patients receiving thyroxine as hormone replacement therapy may develop hyperthyroidism. Based on this, it is recommended to monitor thyroid function after starting somatropin therapy, as well as whenever its dose is changed. Lack of adequate treatment for hypothyroidism may prevent optimal results from somatropin treatment.
Formation of antibodies to somatropin
The formation of antibodies to somatropin is possible. A study of the titer of antibodies to somatropin should be carried out in cases where the patient does not respond to therapy.
Insulin sensitivity
Somatropin reduces insulin sensitivity, especially in large doses in patients with high sensitivity, which can cause the development of hyperglycemia in patients with inadequate insulin secretion.
Thus, previously undiagnosed impaired glucose tolerance and diabetes mellitus may be detected.
In all patients receiving somatropin, periodic monitoring of glucose concentrations is necessary, especially in patients at high risk of developing diabetes mellitus: in patients with obesity, Turner syndrome, a family history of diabetes mellitus, when taking corticosteroids or a pre-existing impairment of glucose tolerance. During treatment with somatropin, more careful monitoring is required in patients diagnosed with type 1 or 2 diabetes mellitus or with impaired glucose tolerance (see section "Interaction with other drugs"). In such patients, the need for dose adjustment of hypoglycemic drugs should be assessed when prescribing somatropin.
Scoliosis
Some children during periods of excessively rapid growth (especially children with PWS) may experience progression of scoliosis. During the entire period of treatment with somatropin, monitoring should be carried out to identify signs of scoliosis. However, available evidence suggests that somatropin therapy does not affect the incidence or severity of scoliosis.
Pancreatitis
Compared with adults, pediatric patients receiving somatropin therapy may have an increased risk of developing pancreatitis. Despite the rarity of this complication, increased attention should be paid to pediatric patients with abdominal pain.
Obesity
Obese patients are more likely to experience adverse events when administered doses based on body weight.
Hyperestrogenism in women
Women with hyperestrogenism or women taking oral estrogens may need higher doses of somatropin than men.
Elderly age
Elderly patients may be more sensitive to the effects of somatropin and, therefore, the likelihood of developing side effects increases. Therefore, it is advisable to use a lower initial dose and a slower increase in the dose of the drug.
There is no experience in treating patients over 60 years of age with somatropin.
Urgent conditions
Safety of continued somatropin therapy in patients with severe illness associated with complications from open cardiac or abdominal surgery, multiple accident-related injuries, and patients with acute respiratory failure receiving replacement therapy for registered indications who are in the process of therapy the above-mentioned diseases appeared, not established. Therefore, the balance of potential risks and benefits of continuing somatropin therapy in patients in an urgent state must be carefully assessed.