Isoprinosine
Immunostimulating agent with nonspecific antiviral effect. It is a complex containing inosine and a salt of 4-acetamidobenzoic acid with N,N-dimethylamino-2-propanol in a molar ratio of 1:3. The effectiveness of the complex is determined by the presence of inosine; the second component increases its availability to lymphocytes. Restores the functions of lymphocytes in conditions of immunosuppression, increases blastogenesis in the population of monocytic cells, stimulates the expression of membrane receptors on the surface of T-helper cells, prevents a decrease in the activity of lymphocyte cells under the influence of glucocorticoids, and normalizes the inclusion of thymidine in them. Isoprinosine has a stimulating effect on the activity of cytotoxic T-lymphocytes and natural killer cells, the functions of T-suppressors and T-helpers, increases the production of IgG, interferon gamma, interleukins (IL)-1 and IL-2, reduces the formation of pro-inflammatory cytokines - IL-4 and IL-10 potentiates the chemotaxis of neutrophils, monocytes and macrophages.
Shows antiviral activity in vivo against Herpes simplex viruses, cytomegalovirus and measles virus, human T-cell lymphoma virus type III, polioviruses, influenza A and B, ECHO virus (human enterocytopathogenic virus), encephalomyocarditis and equine encephalitis. The mechanism of antiviral action is associated with inhibition of viral RNA and the enzyme dihydropteroate synthetase, which is involved in the replication of some viruses. Enhances the synthesis of lymphocyte mRNA suppressed by viruses, which is accompanied by suppression of the biosynthesis of viral RNA and translation of viral proteins, increases the production of alpha and gamma interferons by lymphocytes, which have antiviral properties
Reduces the clinical manifestations of viral diseases, accelerates convalescence, and increases the body's resistance.
When used as an auxiliary drug for infectious lesions of the mucous membranes and skin caused by the Herpes simplex virus, the affected surface heals more quickly than with traditional treatment. New blisters, swelling, erosions and relapses of the disease occur less frequently. With timely use of inosine pranobex, the incidence of viral infections decreases, the duration and severity of the disease decreases.
Pharmacokinetics
After oral administration, it is quickly and almost completely (> 90%) absorbed and has good bioavailability. When taken orally at a dose of 1500 mg, the Cmax of inosine pranobex is achieved after 1 hour and is 600 mcg/ml. Not detectable in the blood 2 hours after administration.
Inosine pranobex consists of inosine and the salt of p-acetamidobenzoic acid with N,N-dimethylamino-2-propanol. Each of the components of inosine pranobex is rapidly metabolized. Almost 100% of metabolites are detected in urine within 8 to 24 hours after administration. Inosine undergoes metabolism in a cycle typical of purine nucleotides, producing uric acid, the concentration of which in the blood serum may increase. As a result, uric acid crystals may form in the urinary tract. The increase in uric acid concentration is nonlinear and can vary by ±10% within 1-3 hours after ingestion. As a result of the metabolism of p-acetamidobenzoic acid, o-acyl glucuronide is formed; N,N-dimethylamino-2-propanol is metabolized to N-oxide. AUC of p-acetamidobenzoic acid >88%, AUC of N,N - dimethylamino-2-propanol >77%. No accumulation was detected in the body.
Inosine and its metabolites are excreted in the urine. When equilibrium concentration is reached when taking a daily dose of 4 g, the daily urinary excretion of p-acetamidobenzoic acid and its metabolite is approximately 85% of the dose taken; T1/2 - 50 min. T1/2 N,N-dimethylamino-2-propanol - 3-5 hours.
Inosine pranobex and its metabolites are completely eliminated from the body within 48 hours.
One of the important medical and social problems of domestic healthcare is the high incidence of influenza and other acute respiratory viral infections (ARVI), caused by more than 250 viruses. During influenza epidemics, especially those caused by new varieties of viruses, a significant number of patients with severe and complicated forms of infection are registered. Mortality from influenza among hospitalized adult patients, despite certain successes in intensive care, remains quite high (up to 2.5%) [1]. Every year, 27.3–41.2 million cases of ARVI are registered in Russia. The cost of treating influenza and its complications in the world annually amounts to about 14.6 billion dollars. In Russia, economic losses from influenza per year are estimated at 10 billion rubles. [2, 3].
All these circumstances require the timely implementation of a set of organizational, anti-epidemic, preventive and emergency treatment measures. However, today, making a correct diagnosis and identifying the causative agent of ARVI on an outpatient basis is difficult.
And the choice of drugs for the treatment and prevention of these diseases is so huge that it gives rise to the problem of polypharmacy, especially in primary healthcare.
In modern medical practice, from the point of view of clinical pharmacology, a simple algorithm for selecting drugs for the treatment of a specific disease is used. There are drugs that affect the cause of the disease (etiotropic treatment); drugs that affect the mechanisms of disease development (pathogenetic treatment), and drugs that affect the symptoms of the disease (symptomatic treatment).
Let us consider from this perspective the well-known groups of drugs for the treatment of influenza and ARVI.
Medicines that suppress various stages of reproduction of the influenza virus and other respiratory viruses are classified as etiotropic drugs.
Pathogenetic drugs include interferon preparations (IFN) and its inducers, since they have an indirect etiotropic and immunocorrective effect.
The third group is symptomatic drugs that alleviate or relieve individual symptoms of ARVI (hyperthermia, headache, cough, rhinitis, etc.), includes non-steroidal anti-inflammatory drugs, antihistamines, mucolytics, antitussives, anticongestives and some other drugs.
It is known that therapy for ARVI and influenza must be started as early as possible after the first symptoms appear: all etiotropic drugs are most effective when taken within the first 24–36 hours of illness.
Etiotropic therapy includes blockers of M2 channels of influenza A virus (amantadine, rimantadine) and inhibitors of neuraminidase activity of influenza A and B viruses (oseltamivir, zanamivir).
The action of the antiviral drug is aimed at a specific stage of the viral replication cycle. The main antigens of the influenza virus are hemagglutinin and neuraminidase. The most significant contribution to the pathogenicity of influenza A viruses is made by hemagglutinin proteins - PB1, PB2, NS1. In addition, 2 proteins (neuraminidase, M2) are associated with the development of viral resistance to antiviral drugs. Treatment of patients with influenza presents certain difficulties due to such disadvantages of antiviral drugs as the formation of resistance of virus strains, maintaining the therapeutic effect only while taking the drug, adverse reactions, etc. [4, 5].
M2 channel blockers (amantadine, rimantadine and adapromine) block M2 protein ion channels and inhibit early viral replication. These drugs are effective against influenza caused by the A virus and are still used successfully in our country. The main disadvantage of this group of drugs is the selective inhibition of the influenza A virus (influenza B virus does not contain the M2 protein). In practice, doctors prescribe M2 channel blockers without clear indications (for the treatment of influenza caused by the B virus or other acute respiratory infections), they are widely advertised, and the drugs are freely sold in pharmacies. All this has led to the development of drug resistance. It is also necessary to take into account that when prescribing M2 channel blockers to elderly patients with concomitant kidney and liver diseases, adjustment of the daily dose is necessary. All this limits the use of these drugs, despite their affordable price.
Neuraminidase inhibitors (zanamivir and oseltamivir) are highly effective in the treatment of influenza caused by both A and B viruses, have a more favorable safety profile than M2 channel blockers, and practically do not cause drug resistance. These drugs block influenza virus neuraminidase, which plays an important role in the release and spread of newly formed virions after the end of the virus replication cycle in the cell - as a result, the ability of the virus to penetrate healthy cells is impaired [6].
Oseltamivir is a prodrug of the powerful influenza virus neuraminidase inhibitor oseltamivir carboxylate. The mechanism of action is based on the conversion of oseltamivir into an active metabolite and blockade of the release of new virions. The drug is easily absorbed from the gastrointestinal tract, the concentration of the active metabolite in plasma reaches its maximum level after 2-3 hours and is at least 75% of the dose taken. The drug, as a rule, is well tolerated; when administered in the first 36 hours of the disease, it shortens the duration of the main symptoms and the disease as a whole. The clinical effectiveness of oseltamivir for influenza has been demonstrated in large multicenter randomized trials. However, the lack of effect on other respiratory viruses (parainfluenza, adenoviruses, respiratory syncytial virus, etc.) and the high cost of the drug limit its widespread use in outpatient practice.
The pathogenetic agents used for influenza and ARVI - IFN preparations and its inducers - are more numerous.
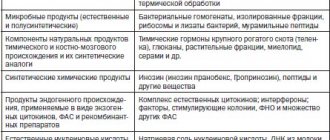
Existing IFN preparations are divided into natural human IFNs of the first generation and recombinant IFNs of the second generation (see table).
IFN inducers approved for clinical use are divided into two main groups: high- and low-molecular-weight, each of which is divided into natural and synthetic drugs. For the prevention and treatment of ARVI, drugs are used that have high interferon-inducing activity, causing the production of different types of IFN in the patient's body. IFN inducers have antiviral, immunocorrective effects and other effects characteristic of IFN [7].
The mechanism of action of this group of drugs is due to nonspecific immunity. The originality of nonspecific mechanisms is determined by the leading role of a number of factors: cellular and tissue immunity, temperature inactivation of viruses, the IFN system, humoral inhibitors of blood serum. Particular importance is attached to the system of mononuclear phagocytes, neutrophils, and natural killer cells. At the same time, various reactions of immunity and nonspecific defense are in coordinated interaction. The state of the T-cell immunity is of great importance in recovery from viral infections. Of particular danger is the ability of a number of viruses to block the primary nonspecific immune response of phagocytes and suppress the synthesis of IFN, which contributes to the deepening of immunodeficiency (suppression of the function of macrophages and neutrophils, blockade of tumor necrosis factor receptors, lymphocytopenia, disruption of the mechanisms of cell differentiation in the system of T- and B-lymphocytes) [ 8, 9].
Severe and complicated forms of influenza and ARVI occur with the development of transient T-cell immunosuppression, a decrease in the functional activity of natural killer cells, the phagocytic and metabolic activity of peripheral blood neutrophils, an increase in the content of serum immune complexes, and the presence of IFN deficiency [10, 11].
With all this, it is necessary to take into account that most drugs from the group of IFN inducers are used only in Russia and have a weak evidence base.
An exception is the drug Isoprinosine (inosine pranobex), which according to the anatomical and therapeutic classification is classified as “other antiviral agents” (ATC code - J05AX05). Isoprinosine has dual – antiviral and immunomodulatory – activity. The drug has been used in medical practice since 1971 for the treatment of cell-mediated immunodeficiency conditions associated with various viral infections (herpes simplex, herpes zoster, cytomegalovirus infection, Epstein-Barr virus).
This molecule was synthesized by P. Gordon (Chicago Medical School, USA) based on the hypothesis that the purine metabolite inosine and its analogues affect mRNA, protein biosynthesis and, due to this, are capable of increasing the adaptive ability of cells to viral infection and their immune defense . Under the name Inosiplex, the drug was patented in the USA in 1972, and by 1990 it was registered and approved for use in more than 70 countries under various trade names in two dosage forms (tablets, syrup), which are currently used as antiviral drugs with immunomodulatory activity [12].
The pharmacokinetic properties of the drug are determined by good absorption from the gastrointestinal tract when taken orally. Cmax in blood plasma is determined after 1–2 hours. The drug is rapidly metabolized similarly to endogenous purine nucleotides with the formation of uric acid and is excreted through the kidneys. Does not accumulate in the body. T1/2 is 3.5 hours for M-M-dimethylamino-2-propranolone and 50 minutes for paracetamidobenzoate. Elimination of the drug and its metabolites from the body occurs within 24–48 hours [13, 14]
The pharmacodynamics of the drug is determined by the ability to inhibit the replication of a wide range of DNA and RNA containing viruses. Isoprinosine has a direct antiviral effect - it suppresses the replication of DNA and RNA viruses by binding to cellular ribosomes and changing their stereochemical structure, as a result of which the synthesis of viral proteins in cells is disrupted and the intensity of virus reproduction is reduced [15]. In addition, the drug supports the functional activity of immunocompetent cells, stimulating the differentiation of T lymphocytes into cytotoxic T cells and T helper cells, and also increases the production of lymphokines, stimulates the production of interleukin-1 (IL-1), IL-2, IFN γ and functional NK cell (natural killer) cell activity. Isoprinosine also potentiates the morphofunctional viability of neutrophils and monocytes, enhancing macrophage chemotaxis and phagocytosis, thereby normalizing cellular immunity. At the same time, by stimulating the differentiation of B lymphocytes into plasma cells and increasing the production of antibodies, it normalizes humoral immunity. In this case, there is an increase in the concentration of immunoglobulins IgG, IgA and IgM, as well as surface markers of complement and virus-neutralizing antibodies [16]. Thanks to the modulation of nonspecific immunity, the body’s resistance to a host of viruses increases [17].
The antiviral activity of Isoprinosine has also been proven against respiratory viruses, including influenza A and B viruses, parainfluenza, adenoviruses, and respiratory syncytial virus [18].
Back in 1974, in studies by DM Pachuta et al. The effectiveness of inosine against rhinovirus infection was established [19]. Many studies confirm the effectiveness of the drug against influenza viruses [20, 21].
The activity of Isoprinosine is equally high both in the first hours of the disease and in the following days [22].
The administration of Isoprinosine in cases of prolonged post-viral asthenia helps to normalize energy metabolism in cells. Penetrating into cells, the drug increases their energy level, enhances resistance to the cytopathogenic effects of viruses and prevents the development of post-viral asthenia syndrome, especially characteristic of influenza and repeated acute respiratory viral infections [23].
The effectiveness and safety of Isoprinosine from the standpoint of evidence-based medicine has been confirmed by foreign and domestic studies.
The use of Isoprinosine for influenza and ARVI reduces the duration and severity of symptoms of the disease (febrile period, intoxication, catarrhal symptoms in the nasopharynx). A decrease in the duration of the disease and relief of symptoms of the disease 48–72 hours after the administration of the drug to children aged from one month to 12 years with ARVI (rhinopharyngitis, acute nasopharyngitis) have been shown; in treated patients the temperature was low and lasted no more than 2 days, general symptoms disappeared within 1–2 days [24].
The use of Isoprinosine for the treatment of ARVI in patients with weakened immune systems reduces the need for antibiotics, does not require additional prescription of other drugs, and shortens the length of hospitalization [24, 25].
A meta-analysis of the clinical and immunological effectiveness of Isoprinosine for respiratory infections in immunocompromised patients, according to published studies in which the drug was used for therapeutic and prophylactic purposes in 2.5 thousand patients (children and adults), also confirmed the effectiveness of therapy [25].
A review of clinical studies of the drug Isoprinosine revealed no serious adverse events. The most common side effects include complaints from the gastrointestinal tract (slight pain in the upper abdomen, nausea, vomiting). [17].
In addition, in studies by RC Batterman (1970) [26], HW Elliott et al. (1972) when using the drug, an increase in uric acid levels was observed [27]. It should be noted, however, that in these studies mean serum uric acid concentrations increased but remained within the normal range (8 mg/100 ml or 480 μmol/L in men). Serum urate concentrations increased from 4.7 mg to 6.3 mg/100 ml after one week of treatment with the drug at a dose of 1 g/day, to 6.6 mg/100 ml after one week of treatment with the drug at a dose of 2 g/day. 7.0 mg/100 ml after a week's course at a dose of 3 g/day and up to 7.2 mg/100 ml after a week of treatment with Isoprinosine at a dose of 4 g/day. After discontinuation of therapy with this drug, serum and urine uric acid levels returned to pretreatment levels. Post-marketing surveillance over many years has not revealed any risk factors associated with the effect of Isoprinosine on serum uric acid levels.
Thus, the drug Isoprinosine, which has dual - antiviral and immunomodulatory - activity, and has a favorable safety profile, confirmed by clinical studies not only in Russia but also abroad, can be recommended for widespread use in primary healthcare as a drug of choice for the treatment of not only influenza , but also ARVI.