Eralfon®
Before and after starting therapy with Eralfon®, blood pressure must be adequately monitored. Eralfon® should be used with caution in the presence of untreated or poorly controlled arterial hypertension. During therapy with Eralfon®, antihypertensive therapy may be necessary. If it is impossible to reduce blood pressure with antihypertensive drugs, therapy with Eralfon® should be discontinued.
A hypertensive crisis accompanied by encephalopathy and convulsions, requiring immediate medical intervention, can also occur during therapy with Eralfon in patients who previously had normal or low blood pressure. Particular attention should be paid to sudden shooting migraine-like headaches as a possible signal of the onset of a crisis (see “Side Effects”).
Epoetin alfa should also be used with caution in patients with chronic liver failure. The safety of epoetin alfa in patients with hepatic impairment has not been established. Due to reduced metabolism in patients with impaired liver function, increased erythropoiesis may occur when using epoetin alfa.
An increased incidence of thrombotic vascular events, such as venous and arterial thrombosis and embolism (including some fatal cases), such as deep vein thrombosis, pulmonary embolism, retinal thrombosis and myocardial infarction, has been observed in patients receiving erythropoietin stimulant drugs.
In addition, cerebrovascular accidents (including cerebral infarction, intracerebral hemorrhage and transient ischemic attacks) have been observed. The reported risk of thrombotic vascular events should be carefully weighed against the benefit of treatment with epoetin alfa, particularly in patients with risk factors. Hemoglobin levels should be closely monitored in all patients due to the potential increased risk of thromboembolic events and deaths observed in patients with elevated hemoglobin levels when treated with Eralfon.
The safety and effectiveness of epoetin alfa therapy in patients with underlying hematological diseases, such as hemolytic anemia, sickle cell anemia, thalassemia, have not been studied.
When treating with Eralfon®, regular monitoring of platelet levels is required, especially during the first 8 weeks, since a dose-dependent relative increase in platelet count may develop, which subsequently normalizes without discontinuing therapy; in rare cases, there is an absolute increase in the platelet count.
Other causes of anemia (iron, folic acid or vitamin B12 deficiency, aluminum toxicity, infection or inflammation, blood loss, hemolysis and bone marrow fibrosis of any origin) should be assessed and treated before initiating epoetin alfa therapy or when deciding to increase the dose. . In most cases, serum ferritin levels decrease simultaneously with an increase in hematocrit. To achieve an optimal response to epoetin alfa, adequate iron stores should be ensured, with supplemental iron supplementation if necessary:
- In patients with chronic renal failure, supplemental iron supplementation is recommended (elemental iron 200-300 mg/day orally in adults and 100-200 mg/day orally in children) if serum ferritin levels are below 100 ng/ml.
— For patients with cancer, additional intake of iron-containing drugs (elemental iron 200-300 mg/day orally) is recommended if transferrin saturation is less than 20%.
— For patients in the autologous blood collection program, supplemental iron supplementation (elemental iron 200 mg/day orally) is recommended for several weeks before the start of autologous blood collection, in order to achieve large iron stores before starting epoetin alfa therapy, as well as throughout the course of epoetin therapy alpha.
- For patients scheduled for major elective orthopedic surgery, additional iron supplementation (elemental iron 200 mg/day orally) is recommended throughout the course of epoetin alfa therapy. If possible, the use of iron supplements should be started prior to initiation of epoetin alfa therapy to ensure adequate iron stores are established.
Very rarely, exacerbations of porphyria were observed early in treatment in patients treated with epoetin alfa. Epoetin alfa should be used with caution in patients with porphyria.
Erythropoiesis-stimulating agents are not necessarily equivalent. Therefore, it should be clarified that patients should be transferred from one erythropoiesis-stimulating drug (such as Eralfon®) to another only with the approval of the attending physician.
In patients with chronic renal failure treated with subcutaneous epoetin, antibody-mediated true erythrocyte aplasia (IEA) was observed very rarely after months and years of treatment.
Rare cases of this disease have also been reported in patients with hepatitis C treated with interferon and ribavirin when erythropoietin-stimulating drugs were concomitantly treated, so they are not approved for the treatment of anemia associated with hepatitis C.
In patients with chronic renal failure who experience a sudden decline in response, as measured by a decrease in hemoglobin (1-2 g/dL per month) with an increase in transfusion requirements, a reticulocyte count should be performed and assessed for typical causes of non-response (eg, deficiency of iron, folic acid or vitamin B12, aluminum toxicity, infection or inflammation, blood loss, hemolysis and bone marrow fibrosis of any origin).
If the anemia-corrected reticulocyte count (i.e., reticulocyte “index”) is low (<20,000/mm3 or <20,000/μL or <0.5%) and the platelet and white blood cell counts are normal, and if no other reasons for the decreased effect, the level of antibodies to erythropoietin should be determined, and the possibility of a bone marrow examination should be considered to diagnose IEA.
If anti-erythropoietin antibody-mediated IEA is suspected, epoetin alfa therapy should be discontinued immediately. Treatment with any other erythropoietin-stimulating drugs should not be initiated as there is a risk of cross-reaction. If indicated, patients may be given appropriate therapy, such as blood transfusions.
Patients with chronic renal failure during treatment with Eralfon® should regularly measure hemoglobin levels until the hemoglobin level reaches stable values, with periodic monitoring thereafter.
To reduce the risk of hypertension, the rate of increase in hemoglobin levels should be approximately 10 g/l (maximum 20 g/l) per month. The dose should be reduced when the hemoglobin level reaches 120 g/l.
In patients with chronic renal failure, the maintained hemoglobin level should not exceed the upper limit of the specified hemoglobin level range. A hemoglobin level of 130 g/L or higher may result in an increased risk of cardiovascular events, including death.
Patients with chronic renal failure and inadequate hemoglobin response to erythropoietin-stimulating drug therapy may be at even greater risk for cardiovascular events and mortality than other patients. Based on currently available information, use of epoetin alfa in patients prior to dialysis (end-stage renal disease) does not increase the rate of progression of renal disease.
Shunt thrombosis has occurred in hemodialysis patients, particularly in patients with a tendency to hypotension or with complications of arteriovenous fistulas (eg, stenoses, aneurysms, etc.). In these patients, early check of the shunt and prevention of thrombosis by administering, for example, acetylsalicylic acid are recommended.
In some cases, hyperkalemia was observed, although its cause-and-effect relationship with the use of the drug has not been established. Serum electrolytes should be monitored in patients with chronic renal failure. If serum potassium levels are elevated or worsening, then, in addition to appropriate treatment of hyperkalemia, discontinuation of epoetin alfa should be considered until the serum potassium level is adjusted.
Due to an increase in hematocrit during therapy with epoetin alfa, patients on hemodialysis may require an increase in the dose of heparin, otherwise occlusion of the dialysis system may occur.
Some patients with chronic renal failure during treatment with epoetin alfa experienced a resumption of menstruation. The possibility of pregnancy and the need for contraceptive measures should be discussed with the patient before starting therapy.
Erythropoiesis-stimulating agents are growth factors that primarily stimulate the formation of red blood cells. Erythropoietin receptors can be expressed on the surface of many tumor cells. As with all growth factors, there are concerns about whether erythropoiesis-stimulating agents can stimulate tumor growth.
In view of the above, the decision to administer a recombinant erythropoietin drug should be made on the basis of a risk-benefit assessment involving the individual patient, and should take into account the specific clinical situation. Factors that should be considered in this assessment include: tumor type and stage; severity of anemia; life expectancy; the setting in which the patient receives treatment; patient preferences.
In patients with cancer receiving chemotherapy, when assessing the suitability of epoetin alfa therapy (particularly in patients at risk of transfusion), the delay of 2-3 weeks between the administration of erythropoiesis-stimulating agents and the appearance of erythropoietin-stimulated red blood cells should be taken into account. If an HIV-infected patient does not respond or responds insufficiently to epoetin alfa therapy, other possible causes of anemia, including iron deficiency, should be considered.
In patients associated with an autologous blood collection program and receiving additional treatment with epoetin alfa, all special warnings and special precautions should be taken into account, especially mandatory volume replacement.
Appropriate blood monitoring standards should always be followed pre- and postoperatively.
Patients scheduled for major elective orthopedic surgery should receive antithrombotic prophylaxis because thrombotic and vascular events may occur in surgical patients, particularly in patients with underlying cardiovascular disease.
In addition, special precautions should be taken in patients with a predisposition to the development of thrombotic complications. Moreover, in patients with a baseline hemoglobin level >130 g/L (8.1 mmol/L), the possibility that treatment with epoetin alfa may be associated with an increased risk of postoperative thrombotic vascular events cannot be excluded. Therefore, in patients with a baseline hemoglobin level > 130 g/l (8.1 mmol/l), the use of epoetin alfa is not recommended.
Eralfon
During treatment with the drug, it is necessary to monitor blood pressure weekly and perform a complete blood count (including platelets, hematocrit, ferritin). In the pre- and postoperative period, Hb should be monitored more often if the initial value was less than 140 g/l.
It must be remembered that the drug in the treatment of anemia does not replace blood transfusion, but reduces the need for its repeated use.
In patients with controlled arterial hypertension or a history of thrombotic complications, an increase in the dose of antihypertensive drugs and anticoagulants may be required, respectively.
When prescribed to patients with liver failure, a slowdown in the metabolism of epoetin alfa and a pronounced increase in erythropoiesis are possible. The safety of the drug in this category of patients has not been established. Although the drug stimulates erythropoiesis, the possibility of the drug affecting the growth of certain types of tumors, incl. bone marrow.
The possibility that a preoperative increase in Hb may predispose to the development of thrombotic complications should be considered. Before undergoing elective surgery, patients should receive adequate prophylactic antiplatelet therapy. In the pre- and postoperative period, it is not recommended to prescribe it to patients with an initial Hb of more than 150 g/l. In adult patients with chronic renal failure and clinically significant ischemic heart disease or heart failure, Hb should not exceed 100-120 g/l.
Before starting treatment, possible causes of an inadequate reaction to the drug should be excluded (deficiency of Fe, folic acid, cyanocobalamin, severe Al3+ poisoning, concomitant infections, inflammatory processes and injuries, hidden bleeding, hemolysis, bone marrow fibrosis of various etiologies) and, if necessary, adjust the treatment. Before starting treatment, iron reserves in the body should be assessed. In most patients with chronic renal failure, in cancer and HIV-infected patients, the plasma ferritin concentration decreases simultaneously with an increase in hematocrit. Ferritin concentrations must be determined throughout the course of treatment. If it is less than 100 ng/ml, replacement therapy with oral Fe preparations is recommended at a rate of 200-300 mg/day (100-200 mg/day for children). Patients donating autologous blood and in the pre- or postoperative period should also receive an adequate amount of Fe orally at a dose of 200 mg/day.
In patients with chronic renal failure, correction of anemia may result in improved appetite and increased absorption of K+ and proteins. Periodic adjustments of dialysis parameters may be required to maintain BUN, creatinine, and K+ concentrations within normal limits. In patients with chronic renal failure, it is necessary to monitor serum electrolytes.
According to available data, the use of the drug in predialysis patients does not accelerate the progression of chronic renal failure. Due to increased hematocrit, it is often necessary to increase the dose of heparin during hemodialysis. With inadequate heparinization, blockage of the dialysis system and thrombosis of shunts are possible, especially in patients with a tendency to hypotension or with complications of an arteriovenous fistula (stenosis, aneurysm, etc.). In such patients, early revision of the shunt and timely prevention of thrombosis (for example, ASA) are recommended.
Use during pregnancy and lactation is possible only in cases where the potential benefit to the mother outweighs the potential risk to the fetus. It is not known whether the drug is excreted in breast milk. When used in women of reproductive age with anemia due to chronic renal failure, menstruation may resume. The patient should be warned about the possibility of pregnancy and the need to use reliable methods of contraception before starting therapy.
Experimental studies on rats and rabbits did not reveal a teratogenic effect when administered intravenously in doses up to 500 IU/kg body weight per day; at higher doses, a weak, statistically insignificant decrease in fertility was noted.
A study of chronic toxicity (in rats and dogs) of epoetin alfa revealed the development of subclinical fibrosis of bone marrow tissue. During the 13-week study, dogs treated subcutaneously or intravenously at doses of 80, 240, or 520 units/kg/day developed anemia without or with signs of bone marrow hypoplasia. Due to the fact that the drug is a human glycoprotein, it is recognized that these changes could be caused by the action of antibodies to epoetin alfa. Similar phenomena were observed in isolated cases when using the drug in veterinary practice and were explained by the appearance of antibodies to epoetin alfa. No carcinogenicity studies have been conducted. The drug does not cause gene mutations in bacteria (Ames test), chromosomal aberrations in mammalian cells, micronucleia in mice, or gene mutations in the HGRT locus.
During the treatment period, until the optimal maintenance dose is established for patients with chronic renal failure, care must be taken when driving vehicles and engaging in other potentially hazardous activities that require increased concentration and speed of psychomotor reactions (increased risk of increased blood pressure at the beginning of therapy).
Patients can be transferred from one stimulant to another only in agreement with the attending physician.
Eralfon®
Treatment of anemia in patients with chronic renal failure
Adult patients undergoing hemodialysis
Eralfon® is administered subcutaneously or intravenously at the end of the dialysis session. When changing the method of administration, the drug is administered at the same dose, then the dose is adjusted if necessary (with the subcutaneous route of administration of the drug, to achieve the same therapeutic effect, a dose of 20-30% less is required than with intravenous administration). Treatment with the drug includes two stages:
1. Correction stage
: with subcutaneous administration of the drug, the initial single dose is 30 IU/kg 3 times a week. When administered intravenously, the initial single dose is 50 IU/kg 3 times a week. The correction period lasts until the optimal level of hemoglobin (100-120 g/l in adults and 95-110 g/l in children) and hematocrit (30-35%) is achieved. These indicators must be monitored weekly.
The following situations are possible:
1) Hematocrit increases from 0.5 to 1% per week. In this case, the dose is not changed until optimal values are achieved.
2) The rate of increase in hematocrit is less than 0.5% per week. In this case, it is necessary to increase the single dose by 1.5 times.
3) Growth rate of more than 1% per week. In this case, it is necessary to reduce the single dose of the drug by 1.5 times.
4) Hematocrit remains low or decreases. It is necessary to analyze the causes of resistance before increasing the dose of the drug. The effectiveness of therapy depends on a correctly selected individual treatment regimen.
2. Maintenance therapy phase
: to maintain the hematocrit at the level of 30-35%, the dose of the drug used at the correction stage should be reduced by 1.5 times. Then the maintenance dose of the drug is selected individually, taking into account the dynamics of hematocrit and hemoglobin levels.
In children undergoing hemodialysis
,
the initial dose
is 50 units/kg 3 times a week.
If necessary, the single dose is increased once every 4 weeks by 25 units/kg until the optimal hemoglobin concentration is achieved. Maintenance dose
for
children weighing less than 10 kg
- 75-150 IU/kg (average 100 IU/kg) 3 times a week,
10-30 kg
- 60-150 IU/kg (average 75 IU/kg) 3 once a week,
more than 30 kg
- 30-100 units/kg (on average 33 units/kg) 3 times a week.
Adult predialysis patients
The initial dose is administered subcutaneously or intravenously 3 times at 50 units/kg per week. If necessary, the single dose is increased once every 4 weeks by 25 units/kg until the optimal hemoglobin concentration is achieved. Maintenance dose - 17-33 IU/kg 3 times a week.
Prevention and treatment of anemia in patients with solid tumors
Before starting treatment, it is recommended to determine the level of endogenous erythropoietin. When the concentration of serum erythropoietin is less than 200 IU/ml, the initial dose of the drug for intravenous administration is 150 IU/kg 3 times a week. If after 4 weeks of treatment the hemoglobin level has increased and is at least 10 g/l or the reticulocyte count has increased by more than 40,000 cells/μl above the initial level, then the dose of the drug remains the same (150 IU/kg body weight 3 times a week).
If after 4 weeks of treatment the increase in hemoglobin level is less than 10 g / l and the increase in the number of reticulocytes is less than 40,000 cells / μl compared to the initial level, then over the next 4 weeks the dose is increased to 300 IU / kg body weight 3 times a week . If after an additional 4 weeks of treatment at a drug dose of 300 IU/kg, the hemoglobin level has increased and is at least 10 g/l or the reticulocyte count has increased by more than 40,000 cells/μl, then the existing dose of the drug is maintained (300 IU/kg body weight 3 once a week). If after 4 weeks of treatment at a dose of 300 IU/kg body weight, the hemoglobin level increases by less than 10 g/L and the increase in the reticulocyte count is less than 40,000 cells/μL compared to the baseline level, treatment should be discontinued. If the hemoglobin level increases by more than 20 g/l within a month, the dose of the drug must be reduced by 25%. If the hemoglobin level exceeds 140 g/l, it is necessary to suspend treatment until the hemoglobin level decreases below 120 g/l and then continue administration of the drug at a dose 25% lower than the original one. Therapy with the drug should continue for one month after the end of chemotherapy.
Serum ferritin levels (or serum iron levels) should be determined in all patients before and during treatment with the drug. If necessary, additional iron intake is prescribed.
Prevention and treatment of anemia in patients with HIV infection
It is recommended to determine the initial level of endogenous erythropoietin in the blood serum before starting treatment with Eralfon®. Studies have shown that if the level of erythropoietin is more than 500 IU/ml, the effect of drug therapy is unlikely.
1. Correction stage
: the drug is prescribed at a dose of 100 IU/kg 3 times a week subcutaneously or intravenously for 8 weeks. If after 8 weeks of therapy it has not been possible to achieve a satisfactory effect (for example, to reduce the need for blood transfusions or to achieve an increase in hemoglobin levels), the dose can be gradually increased (no more than once every 4 weeks) by 50-100 IU/kg 3 times week. If it was not possible to achieve a satisfactory effect from therapy with Eralfon® at a dose of 300 IU/kg 3 times a week, then a response to further therapy at higher doses is unlikely.
2. Maintenance therapy stage:
after achieving a satisfactory effect in the anemia correction phase, the maintenance dose should ensure a hematocrit level within 30-35%, depending on changes in the dose of zidovudine, the presence of concomitant infectious or inflammatory diseases. If the hematocrit is more than 40%, the drug should be discontinued until the hematocrit decreases to 36%. When restarting therapy, the dose of epoetin alfa should be reduced by 25%, followed by adjustments to maintain the required hematocrit level. Serum ferritin levels (or serum iron levels) should be determined in all patients before and during treatment with the drug. If necessary, additional iron intake is prescribed.
Prevention and treatment of anemia in patients with multiple myeloma, low-grade non-Hodgkin lymphomas and chronic lymphocytic leukemia
In these patients, the advisability of treatment with epoetin alfa is determined by the inadequate synthesis of endogenous erythropoietin against the background of the development of anemia. When hemoglobin levels are below 100 g/l and serum erythropoietin levels are below 100 IU/ml, Eralfon® is administered subcutaneously at a starting dose of 100 IU/kg three times a week.
Laboratory monitoring of hemodynamic parameters is carried out weekly. If necessary, the dose of the drug is adjusted upward or downward every 3-4 weeks. If, upon reaching a weekly dose of 600 IU/kg, an increase in hemoglobin levels is not observed, further use of epoetin alfa should be discontinued as ineffective.
Prevention and treatment of anemia in patients with rheumatoid arthritis
In patients with rheumatoid arthritis, suppression of the synthesis of endogenous erythropoietin is observed under the influence of increased concentrations of anti-inflammatory cytokines. Treatment of anemia in these patients is carried out with the drug when administered subcutaneously at a dose of 50-75 IU/kg 3 times a week. If the hemoglobin level increases by less than 10 g/l after 4 weeks of treatment, the dose of the drug is increased to 150-200 IU/kg 3 times a week. Further increase in dose seems inappropriate.
Treatment and prevention of anemia in premature infants born with low birth weight
Eralfon® is administered subcutaneously at a dose of 200 IU/kg three times a week, starting from the 6th day of life, until the target levels of hemoglobin and hematocrit are achieved, but not more than 6 weeks.
Adult patients participating in a pre-surgical autologous blood collection program
It is recommended to use intravenous administration of the drug. Epoetin alfa should be administered at the end of the blood collection procedure. Before prescribing the drug, all contraindications to autologous blood collection should be taken into account. Before surgery, Eralfon® should be prescribed 2 times a week for 3 weeks. At each doctor's visit, a portion of blood is taken from the patient (if hematocrit >33% and/or hemoglobin level >110 g/l) and stored for autologous transfusion. The recommended dose of Eralfon® is 600 IU/kg body weight 2 times a week. Serum ferritin levels (or serum iron levels) should be determined in all patients before and during treatment with the drug. If necessary, additional iron intake is prescribed.
If anemia is present, the cause should be determined before initiating epoetin alfa therapy. It is necessary to ensure adequate intake of iron into the body as soon as possible by prescribing an oral iron supplement at a dose of 200 mg/day (calculated as ferrous iron) and maintain iron intake at this level throughout the course of therapy.
Patients in the pre- and postoperative period who are not participating in the autologous blood collection program
It is recommended to use subcutaneous administration of the drug at a dose of 600 IU/kg body weight per week for 3 weeks preceding surgery (21st, 14th and 7th days before surgery) and on the day of surgery. If necessary, when for medical reasons it is necessary to shorten the preoperative period, Eralfon® can be prescribed daily at a dose of 300 IU/kg body weight for 10 days before surgery, on the day of surgery and for 4 days after surgery. If the preoperative hemoglobin level reaches 150 g/L or higher, the use of epoetin alfa should be discontinued. Before initiating therapy with epoetin alfa, it is necessary to ensure that patients do not have iron deficiency. All patients should receive adequate amounts of iron (orally 200 mg/day based on ferrous iron) throughout the course of treatment. If possible, supplemental oral iron intake should be provided prior to initiation of epoetin alfa therapy to ensure adequate iron storage in the patient.
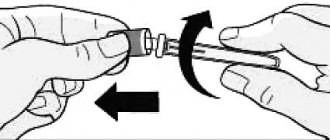
How the syringe works
with automatic needle protection device:
Once the injection is complete, the needle and syringe will move back into the safety device. Attention! Avoid contact with the clamps while preparing the syringe! The device is activated by pressing the rod down to the clamps. Carefully inspect the prefilled glass syringe with safety device. Remove the protective cap from the needle. Inject according to standard procedure. Press the rod with your thumb and hold until the entire dose of the drug has been administered. The safety device is not activated until the entire dose of the drug has been administered. Remove the needle, release the stem, and allow the guard to move forward until the needle is completely protected and locked in place.
with non-automatic needle protection device:
administer the injection according to standard procedure. Attention! When injecting, keep your fingers on the safety guard to prevent premature activation of the safety device. After injection, move the guard along the needle. An audible click will indicate the correct action. During the entire procedure, your fingers should be behind the needle.
Eralfon solution d/iv and subcutaneous injection 10 thousand IU 1 ml No. 1
3 years. Do not use after the expiration date indicated on the package.
special instructions
- Use with caution in patients with a history of convulsive reactions; Patients at increased risk of developing thrombosis or other vascular complications require careful medical monitoring.
- Use with caution for gout.
- Before use, ensure that patients with arterial hypertension have received effective antihypertensive therapy.
- During use, it is necessary to monitor blood pressure levels, paying attention to the occurrence or worsening of unusual headaches. This may require adjustment of therapy or prescription of antihypertensive drugs. If blood pressure does not decrease despite adequate therapy, epoetin alfa should be discontinued.
- Before starting the use of epoetin alfa, the state of the iron depot in the body should be assessed. In most patients with chronic renal failure, in cancer and HIV-infected patients, the level of ferritin in the blood plasma decreases simultaneously with an increase in hematocrit. Ferritin levels must be determined throughout the course of treatment. If it is less than 100 ng/ml, iron replacement therapy is recommended. Patients who donate autologous blood and are in the pre- or postoperative period should also receive additional adequate amounts of iron.
- During the period of use, hemoglobin levels should be monitored at least once a week until a stable level is reached, then somewhat less frequently. In the pre- and postoperative period, hemoglobin levels should be checked more often if the initial level was less than 14 g/dL. Hematocrit levels should also be monitored regularly. During the first 8 weeks of therapy, the platelet count should be regularly monitored because epoetin alfa can cause a moderate dose-dependent increase in the number of platelets, which independently returns to normal during the course of therapy; thrombocytosis develops rarely.
- It should be taken into account that a preoperative increase in hemoglobin levels may serve as a predisposing factor to the development of thrombotic complications. Patients should receive adequate prophylactic antithrombotic therapy before undergoing elective surgery.
- In the pre- and postoperative period, it is not recommended to use epoetin alfa if the initial hemoglobin level is more than 15 g/dl.
- Use with caution in patients with porphyria. In case of chronic renal failure during therapy with epoetin alfa, an exacerbation of porphyria is possible.
- Correction of anemia may be accompanied by improved appetite and increased absorption of potassium and proteins. It should be borne in mind that it may be necessary to periodically adjust dialysis parameters to maintain levels of urea, creatinine and potassium within normal limits. In patients with chronic renal failure, it is necessary to monitor the level of electrolytes in the blood serum.
- Hemodialysis patients treated with epoetin alfa often require increased heparin dosage during dialysis due to increased hematocrit. With an inadequate dose of heparin, occlusion of the dialysis system is possible.
- In patients with chronic renal failure and clinically significant coronary artery disease or congestive heart failure, maintenance hemoglobin levels should not exceed the upper limit of the optimal recommended level (no more than 10-12 g/dL in adults).
- When used in patients with impaired liver function, the biotransformation of epoetin alfa may be slowed down and erythropoiesis may be significantly increased. The safety of epoetin alfa in this category of patients has not been established.
- The possibility that epoetin alfa may influence the growth of certain types of tumors, especially bone marrow malignancies, cannot be completely excluded.
- All special warnings and precautions associated with the autologous blood collection program should be observed (this applies to all patients receiving epoetin alfa).
- The therapeutic effectiveness of epoetin alfa may be reduced by deficiency of iron, folic acid, vitamin B12, aluminum intoxication, intercurrent diseases, hidden bleeding, hemolysis, bone marrow fibrosis.
- In experimental animal studies examining the chronic toxicity of epoetin alfa, subclinical fibrosis of bone marrow tissue, as well as anemia with or without signs of bone marrow hypoplasia, were observed in some cases. It is believed that this is due to the appearance of antibodies to epoetin alfa.
- No mutagenic effect was detected.
Description
Hematopoiesis stimulator.
Dosage form
The solution for intravenous and subcutaneous administration is transparent, colorless.
Use in children
In children undergoing hemodialysis, the initial dose is 50 units/kg 3 times a week. If necessary, the single dose is increased once every 4 weeks by 25 units/kg until the optimal hemoglobin concentration is achieved. Maintenance dose in children weighing less than 10 kg - 75-150 IU/kg (average 100 IU/kg), 10-30 kg - 60-150 IU/kg (average 75 IU/kg), more than 30 kg - 30-100 units/kg (average 33 units/kg).
Action
Recombinant human erythropoietin is a purified glycoprotein. Stimulates erythropoiesis. It is synthesized in mammalian cells into which the gene encoding human erythropoietin is embedded. Biological and immunological properties are identical to human erythropoietin isolated from urine. The synthesis of endogenous erythropoietin occurs in the kidneys and depends on the level of blood oxygenation.
Side effects
Flu-like syndrome: possible dizziness, drowsiness, fever, headache, pain in joints and muscles (mainly at the beginning of treatment).
From the cardiovascular system: a dose-dependent increase in blood pressure is possible; worsening of arterial hypertension (most often in patients with chronic renal failure); in some cases - hypertensive crises, malignant arterial hypertension with symptoms of encephalopathy (headache, confusion) and generalized tonic-clonic seizures.
From the hematopoietic system: rarely - thrombocytosis.
From the coagulation system: in some cases - shunt thrombosis (in patients on hemodialysis with a tendency to hypotension or in the presence of stenoses, aneurysms).
From the urinary system: possible hyperkalemia, hyperphosphatemia, increased concentrations of urea, creatinine, uric acids in the blood plasma (in patients with chronic renal failure).
Allergic reactions: in some cases - mild or moderate skin rash, eczema, urticaria, itching, angioedema.
Local reactions: possible redness, burning sensation, mild or moderate pain at the injection site (more often occurs with subcutaneous administration).
Other: rarely - potentially serious complications associated with respiratory failure or decreased blood pressure; immune reactions (has minimal ability to induce the formation of antibodies).
Use during pregnancy and breastfeeding
The safety of epoetin alfa during pregnancy and lactation has not been established.
Interaction
The effect of epoetin alfa may be enhanced by concomitant administration of blood products.
With simultaneous use of epoetin alfa with cyclosporine, a decrease in the concentration of the latter in plasma is possible due to an increase in the degree of its binding to erythrocytes (when using this combination, it is necessary to control the concentration of cyclosporine in plasma and, if necessary, increase its dose).
Epoetin alfa should not be mixed with solutions of other drugs.
Overdose
Symptoms: increased side effects.
Treatment: symptomatic. If the level of hemoglobin is high - bloodletting.
Impact on the ability to drive vehicles and operate machinery
When using epoetin alfa, until the optimal maintenance dose is established, patients with chronic renal failure should avoid engaging in potentially hazardous activities due to an increased risk of developing arterial hypertension at the beginning of therapy.