Pharmacological properties of the drug Recormon
Pharmacodynamics. Epoetin beta is a glycoprotein, a factor stimulating mitosis and a hormone of cell differentiation, stimulates the formation of red blood cells from progenitor cells (a class of stem cells). Recombinant epoetin beta, obtained by genetic engineering, is identical in its amino acid and carbohydrate composition to erythropoietin isolated from the urine of patients with anemia. Epoetin beta has a high degree of purification, which corresponds to modern technological capabilities. In particular, the active substance that is administered to a person does not contain remnants of the cell lines used to obtain the drug. Epoetin beta after intravenous and subcutaneous administration increases the number of red blood cells, reticulocytes and the level of hemoglobin in the blood, as well as the rate of incorporation of 59Fe into cells, and does not affect leukopoiesis. No cytotoxic effect of erythropoietin on human bone marrow or skin cells has been detected. Erythropoietin is a growth factor that primarily stimulates red blood cell production, but erythropoietin receptors can also be expressed on the surface of various tumors. There is insufficient information whether epoetin drugs affect the acceleration of tumor progression and survival. There have been two robust studies of the effect of epoetins on survival and/or tumor progression in patients with high hemoglobin levels. In a randomized, placebo-controlled trial, epoetin alfa was used in 939 patients with breast cancer and hemoglobin levels of 12–14 g/dL. After 4 months, an increase in mortality was observed from 3 to 6% in women who received epoetin alfa. Another placebo-controlled study used epoetin beta in 351 patients with head and neck cancer and hemoglobin levels of 14 g/dL in women and 15 g/dL in men. At the same time, survival was recorded in patients who used epoetin beta. The results of these studies are questioned because there is an imbalance between the study groups, especially in terms of tumor location, heterogeneity of the study population, and the presence of aggravating factors, such as smoking. Results from several other studies show a trend toward improved survival with epoetins, suggesting no adverse effect on tumor progression. In rare isolated cases, neutralizing antibodies to epoetin beta may be formed with or without the development of red cell aplasia of the bone marrow. Pharmacokinetics. In healthy individuals and in patients with uremia, the half-life of epoetin beta with intravenous administration is 4–12 hours. The volume of distribution is equal to or 2 times the volume of circulating plasma. With subcutaneous administration of the drug to patients with uremia, prolonged absorption ensures a plateau phase of drug concentration in the blood serum, and the maximum concentration is achieved on average 12–28 hours after administration. With this route of administration, the half-life in the terminal phase is longer than after intravenous administration, averaging 12–28 hours. The bioavailability of epoetin beta with subcutaneous administration is 23–42%.
Recormon, 2000 IU, solution for intravenous and subcutaneous administration, 0.3 ml, 6 pcs.
Treatment of anemia in patients with chronic renal failure
PC
or
IV
for 2 minutes. For patients on hemodialysis - through an arteriovenous shunt at the end of the dialysis session. For patients not receiving hemodialysis, it is preferable to administer the drug subcutaneously to avoid puncture of peripheral veins.
The goal of treatment is a hematocrit level of 30–35% or eliminating the need for blood transfusions. The weekly increase in hematocrit should not exceed 0.5%. It should not exceed more than 35%. In patients with arterial hypertension, cardiovascular and cerebrovascular diseases, the weekly increase in hematocrit and its target values should be determined individually, depending on the clinical picture. For some patients, the optimal hematocrit is below 30%.
Treatment with Recormon® is carried out in 2 stages.
Correction stage.
SC, initial dose 20 IU/kg 3 times a week. If the increase in hematocrit is insufficient (less than 0.5% per week), the dose can be increased every 4 weeks by 20 IU/kg 3 times a week. The total weekly dose of the drug can also be divided into daily administrations.
IV at an initial dose of 40 IU/kg 3 times a week. If the hematocrit does not increase sufficiently after a month, the dose can be increased to 80 IU/kg 3 times a week. If necessary, the dose should be increased in the future by 20 IU/kg 3 times a week at monthly intervals.
Regardless of the route of administration, the maximum dose should not exceed 720 IU/kg per week.
Maintenance therapy.
To maintain the hematocrit at 30–35%, the dose should first be reduced by 2 times from the previous dose. Subsequently, the maintenance dose is selected individually, at intervals of 1 or 2 weeks. For subcutaneous administration, the weekly dose can be administered in one dose or divided into 3 or 7 administrations per week. When the condition stabilizes against the background of a single administration per week, you can switch to a single administration at a two-week interval; in this case, an increase in the dose may be necessary.
In children, the dose of the drug depends on age: as a rule, the younger the child, the higher doses of epoetin beta he needs. But since it is impossible to predict the individual response to the drug, it is advisable to start with the recommended dosage regimen.
Treatment with Recormon® is usually long-term. If necessary, it can be interrupted at any time.
Prevention of anemia in premature newborns.
PC
250 IU/kg 3 times a week as early as possible, preferably from the 3rd day of life for 6 weeks.
Prevention and treatment of anemia in patients with solid tumors
It is carried out when hemoglobin before the start of chemotherapy is ≤ 130 g/l. The drug is administered subcutaneously
at an initial dose of 450 IU/kg per week, dividing the weekly dose into 3 or 7 injections. If after 4 weeks the hemoglobin does not increase sufficiently, the dose should be doubled. Treatment is continued for no more than 3 weeks after the end of chemotherapy.
If during the first cycle of chemotherapy, the hemoglobin level, despite treatment with epoetin beta, decreases by more than 10 g/l, further use of the drug may be ineffective.
An increase in hemoglobin by more than 20 g/l per month or above 140 g/l should be avoided. If hemoglobin increases by more than 20 g/l per month, the dose of epoetin beta should be reduced by 50%. If hemoglobin exceeds 140 g/L, the drug is discontinued until hemoglobin decreases to ≤120 g/L, and then treatment is resumed at a dose of half the previous weekly dose.
Treatment of anemia in patients with multiple myeloma, low-grade non-Hodgkin's lymphoma, or chronic lymphocytic leukemia
These patients usually have a deficiency of endogenous erythropoietin. It is diagnosed by the relationship between the degree of anemia and the insufficient concentration of erythropoietin in the serum. Relative erythropoietin deficiency is determined by:
| Hemoglobin, g/l | Serum erythropoietin, IU/ml |
| >90–<100 | ≤100 |
| >80–≤90 | ≤180 |
| ≤80 | ≤300 |
The above parameters should be determined at least 7 days after the last blood transfusion and the last cycle of cytotoxic chemotherapy.
The drug is administered subcutaneously
The recommended starting dose is 450 IU/kg per week. The weekly dose can be administered at one time or divided into 3 or 7 injections. If after 4 weeks the hemoglobin level increases by at least 10 g/l, treatment is continued at the same dose. If after 4 weeks hemoglobin increases by less than 10 g/l, the dose can be increased to 900 IU/kg per week. If after 8 weeks of treatment hemoglobin has not increased by at least 10 g/l, a positive effect is unlikely and the drug should be discontinued.
For chronic lymphocytic leukemia, treatment should be continued until 4 weeks after the end of chemotherapy. The maximum dose is no more than 900 IU/kg per week. If over 4 weeks of treatment hemoglobin increases by more than 20 g/l, the dose of Recormon® should be reduced by half. If hemoglobin exceeds 140 g/l, treatment with Recormon® should be interrupted until its level reaches ≤130 g/l, after which therapy is resumed at a dose equal to 50% of the previous weekly dose.
Treatment should only be restarted if the most likely cause of the anemia is erythropoietin deficiency.
Preparing patients for the collection of donor blood for subsequent autohemotransfusion
IV
or
s/c
2 times a week for 4 weeks. In cases where the patient's hematocrit (≥33%) allows blood sampling, Recormon® is administered at the end of the procedure. Throughout the course of treatment, the hematocrit should not exceed 48%.
The dose of the drug is determined individually by the transfusiologist and the surgeon, depending on the volume of blood that will be taken from the patient and his erythrocyte reserve. The volume of blood that will be taken from the patient depends on the estimated blood loss, available blood conservation techniques and the general condition of the patient; it must be sufficient to avoid a blood transfusion from another donor. The volume of blood that will be taken from the patient is expressed in units (one unit is equivalent to 180 ml of red blood cells).
The possibility of donation depends mainly on the blood volume of a given patient and the initial hematocrit. Both indicators determine the endogenous erythrocyte reserve, which can be calculated using the following formula:
endogenous erythrocyte reserve = blood volume [ml] × (hematocrit − 33) : 100
women: blood volume [ml] = 41 [ml/kg] × body weight [kg] + 1200 [ml]
men: blood volume [ml] = 44 [ml/kg] × body weight [kg] + 1600 [ml] (for body weight ≥45 kg).
Indications for use of Recormon® and its single dose are determined by nomograms, based on the required volume of donor blood and endogenous erythrocyte reserve.
The maximum dose should not exceed 1600 IU/kg per week when administered intravenously and 1200 IU/kg per week when administered subcutaneously.
Instructions for use
A bottle of lyophilized powder for the preparation of solution for injection
The powder is dissolved in the contents of the supplied ampoule with solvent. The prepared solution should be used within 2 hours. Only a light, transparent or slightly opalescent solution that does not contain visible impurities should be used. Unused solution must be destroyed.
To avoid incompatibility or reduced activity of the drug, you should not use a glass syringe; you should use only plastic materials for injection.
Syringe tube
with Recormon® is ready for use. The solution it contains is sterile and does not contain preservatives. Only a light, transparent or slightly opalescent solution that does not contain visible impurities should be used. If after the injection a certain amount of the drug remains in the syringe tube, repeated administration is unacceptable.
Instructions for use of the syringe tube
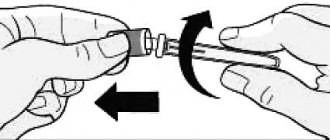
1. Remove one syringe tube from the package and make sure that the solution is transparent, colorless and free of visible impurities. Remove the cap from the syringe.
2. Remove one needle from the package, put it on the syringe and remove the protective cap from the needle.
3. Remove air from the syringe and needle by holding the syringe vertically and carefully pushing the plunger upward. Press the plunger until the required dose of Recormon® remains in the syringe.
4. Wipe the skin at the injection site with cotton wool moistened with alcohol. Using your thumb and index finger, press the skin into a fold. Holding the syringe body close to the needle, insert the needle under the skin. Inject Recormon® solution. Quickly remove the needle and press the injection site with sterile dry cotton wool.
Cartridge
with Recormon® for syringe pen Reko-Pen is a two-section cartridge containing powder for preparing an injection solution and a solvent with preservatives. The finished solution is obtained by inserting the cartridge into the Reko-Pen syringe pen in accordance with the instructions.
Cartridges with Recormon® should only be used in the Reco-Pen syringe pen.
It is recommended to use needles for the Reco-Pen syringe pen (for example, Penfine needles).
The solution prepared in the cartridge is stored for 1 month at a temperature of 2-8 °C.
Indications for use of the drug Recormon
- anemia of renal origin in patients with chronic renal failure in patients on dialysis);
- symptomatic anemia of renal origin in patients who do not undergo dialysis;
- prevention of anemia in premature newborns who were born weighing 750–1500 g before the 34th week of pregnancy;
- symptomatic anemia in patients with solid tumors receiving chemotherapy;
- increasing the volume of autologous blood intended for subsequent autotransfusion to patients who need to avoid blood transfusion.
Recormon solution for intravenous and subcutaneous administration 10000 IU 0.6 ml syringes 6 pcs.
Treatment of anemia in patients with chronic kidney disease. Subcutaneously or intravenously (within 2 minutes). For patients on hemodialysis - through an arteriovenous shunt at the end of the dialysis session. For patients not receiving hemodialysis, it is preferable to administer the drug subcutaneously to avoid puncture of peripheral veins. The goal of treatment is a hemoglobin (Hb) level of 100-120 g/l. Hb should not exceed 120 g/l. If Hb increases by more than 20 g/l (1.3 mmol/l) over 4 weeks, the dose of the drug should be reduced. In patients with arterial hypertension, cardiovascular and cerebrovascular diseases, the weekly increase in Hb and its target values should be determined individually, depending on the clinical picture. The patient should be carefully monitored in order to select the minimum dose sufficient to ensure the maximum effect of the drug. Treatment with Recormon is carried out in 2 stages. Correction stage. Subcutaneously - initial dose - 20 IU/kg 3 times a week. If the increase in Hb is insufficient (less than 2.5 g/l per week), the dose can be increased every 4 weeks by 20 IU/kg 3 times a week. The total weekly dose of the drug can also be divided into daily administrations. Intravenously - initial dose - 40 IU/kg 3 times a week. If there is insufficient increase in Hb after a month, the dose can be increased to 80 IU/kg 3 times a week. If necessary, in the future the dose should be increased by 20 IU/kg 3 times a week, at monthly intervals. Regardless of the route of administration, the maximum dose should not exceed 720 IU/kg per week. Maintenance therapy. To maintain the target Hb value (100-120 g/l), the dose should first be reduced by 2 times from the previous one. Subsequently, the maintenance dose is selected individually, at intervals of 2 or 4 weeks. When administered subcutaneously, the weekly dose can be administered in one dose or divided into 3 or 7 administrations per week. When the condition stabilizes against the background of a single administration per week, you can switch to a single administration at a two-week interval; in this case, an increase in the dose may be necessary. Treatment with Recormon is usually long-term. If necessary, it can be interrupted at any time. Treatment of symptomatic anemia in patients with solid and hematological non-myeloid tumors receiving chemotherapy. The drug is administered subcutaneously at an initial dose of 30,000 IU per week (450 IU/kg per week), a single dose or a weekly dose can be divided into 3 or 7 injections. Therapy with Recormon is indicated for Hb ≥110 g/l (6.83 mmol/l). The Hb value should not exceed 130 g/l (8.07 mmol/l). If Hb increases by 10 g/l (0.62 mmol/l) after 4 weeks, therapy should be continued at the same dose. If Hb increases by less than 10 g/l (0.62 mmol/l) after 4 weeks, the dose should be doubled. If there is no increase in Hb by 10 g/l (0.62 mmol/l) after 8 weeks, treatment should be interrupted, because response to therapy with Recormon® is unlikely. Treatment should be continued for 4 weeks after the end of chemotherapy. The maximum dose should not exceed 60,000 IU per week. When the target Hb value for a particular patient is achieved, the dose of the drug should be reduced by 25-50%. To prevent an increase in Hb above 130 g/l, further dose reduction may be required. If Hb increases by more than 20 g/l (1.3 mmol/l) per month, the dose of Recormon should be reduced by 25-50%. Preparing patients for the collection of donor blood for subsequent autohemotransfusion. Intravenously (over 2 minutes) or subcutaneously, 2 times a week for 4 weeks. In cases where the patient's hematocrit (? 33%) allows blood sampling, Recormon should be administered at the end of the procedure. Throughout the course of treatment, the hematocrit should not exceed 48%. The dose of the drug is determined by the transfusiologist and the surgeon individually, depending on the volume of blood that will be taken from the patient and his erythrocyte reserve: 1. The volume of blood that will be taken from the patient depends on the expected blood loss and available blood preservation techniques and the general condition of the patient; it must be sufficient to avoid a blood transfusion from another donor. 2. The volume of blood that will be taken from the patient is expressed in units (one unit is equivalent to 180 ml of red blood cells). 3. The possibility of donation depends mainly on the blood volume of a given patient and the initial hematocrit. Both indicators determine the endogenous erythrocyte reserve, which is calculated using the following formula: Endogenous erythrocyte reserve = blood volume [ml] x (hematocrit - 33): 100 Women: blood volume [ml] = 41 [ml/kg] x body weight [kg] + 1200 [ml] Men: blood volume [ml] = 44 [ml/kg] x body weight [kg] + 1600 [ml] (with body weight ? 45 kg). The indication for use of the drug Recormon and its single dose are determined by nomograms, based on the required volume of donor blood and endogenous erythrocyte reserve. The maximum dose should not exceed 1600 IU/kg per week when administered intravenously and 1200 IU/kg per week when administered subcutaneously. Prevention of anemia in premature newborns. Subcutaneously, 250 IU/kg 3 times a week, as early as possible, preferably from the 3rd day of life, for 6 weeks. Dosing in special patient groups. Children and teenagers. In children and adolescents, the dose of the drug depends on age: as a rule, the younger the age, the higher doses of Recormon are required. But, since it is impossible to predict the individual response to the drug, it is advisable to start with a standard dosage regimen. When treating anemia associated with chronic kidney disease, Recormon should not be prescribed to children under 2 years of age. Elderly age. In clinical studies, the need for dose changes was not determined. Mode of application. The syringe tube with Recormon is ready for use. The solution it contains is sterile and does not contain preservatives. Only a light, transparent or slightly opalescent solution that does not contain visible inclusions should be used. If after the injection a certain amount of the drug remains in the syringe tube, repeated administration is unacceptable.
Use of the drug Recormon
Treatment of anemia in patients with chronic renal failure The drug is administered subcutaneously or intravenously. When administered intravenously, the drug must be administered over 2 minutes. For patients on hemodialysis, Recormon is administered through an arteriovenous shunt at the end of the dialysis session. For patients who do not undergo hemodialysis, it is better to administer the drug subcutaneously so as not to puncture the peripheral veins. The goal of treatment is to achieve a hematocrit level of 30–35% or eliminate the need for blood transfusions. The weekly increase in hematocrit should not exceed 0.5%. The hematocrit should not exceed 35%. In patients with hypertension (arterial hypertension), other cardiovascular and cerebrovascular diseases, the weekly increase in hematocrit should be determined individually, depending on the clinical picture. For some patients, the optimal hematocrit level may be below 30%. Treatment is carried out in two stages. Initial therapy (correction stage) With subcutaneous administration, the initial dose is 20 IU/kg body weight 3 times a week. The total weekly dose of the drug can also be divided into daily administrations at lower doses. If the hematocrit level does not increase sufficiently (≤0.5% per week), the dose can be increased monthly by 20 IU/kg 3 times a week. When administered intravenously, the initial dose is 40 IU/kg 3 times a week. If the hematocrit does not increase sufficiently after a month, the dose can be increased to 80 IU/kg body weight 3 times a week. If it is necessary to further increase the dose, it is increased by 20 IU/kg 3 times a week at monthly intervals. Regardless of the method of administration, the maximum dose should not exceed 720 IU/kg/week. Maintenance therapy. To maintain the hematocrit at 30–35%, the dose must first be reduced by 2 times relative to the previous one. Over time, the maintenance dose is adjusted individually, at intervals of 1–2 weeks. Treatment with Recormon is usually long-term. If necessary, it can be interrupted at any time. In children, the dose of the drug depends on age; as a rule, the younger the child’s age, the larger the dose of the drug is needed. But since it is impossible to predict the individual reaction to the administration of the drug, it is advisable to start treatment with the recommended dosage regimen. Prevention of anemia in premature newborns The drug is administered subcutaneously at a dose of 250 IU/kg body weight 3 times a week. Treatment with erythropoietin should begin as early as possible, preferably from the 3rd day of life, and continue for 6 weeks. Prevention and treatment of anemia in patients with cancer The drug is administered subcutaneously, dividing the weekly dose into 3 or 7 injections. The recommended starting dose is 30,000 IU per week (450 IU/kg/week). Treatment with Recormon is indicated when the hemoglobin level before the start of chemotherapy is not higher than 11 g/dl. The hemoglobin level should not be higher than 13 g/dl. If after 4 weeks the hemoglobin level increases by at least 1 g/dL, treatment should be continued without changing the dosage. If hemoglobin levels do not increase, the weekly dose of the drug should be doubled. If after 8 weeks of treatment the hemoglobin level does not increase by at least 1 g/day, further use of the drug may be ineffective. It is recommended to continue treatment for another 4 weeks after the end of chemotherapy. The maximum dose of the drug should not exceed 60,000 IU per week. When the required level of hemoglobin, individual for each patient, is achieved, the dose of the drug must be reduced by 25–50%, maintaining hemoglobin at the required level. In the future, if necessary, a dose of the drug should be used so that the hemoglobin level does not exceed 13 g/dl. If hemoglobin increases by more than 2 g/dL in 4 weeks, the dose of the drug should be reduced by 25–50%. Preparing patients for autologous blood collection for subsequent autohemotransfusion The drug is administered 2 times a week for 4 weeks. In cases where the patient's hematocrit allows blood sampling (33%), epoetin beta is administered at the end of the procedure. Throughout the course of treatment, the hematocrit should not exceed 48%. The dose of the drug is determined by the transfusiologist and the surgeon individually, depending on the volume of blood that will be taken from the patient and his erythrocyte reserve. The volume of blood that will be taken from the patient depends on the expected blood loss, the availability of blood conservation techniques and the general condition of the patient; it must be sufficient to avoid a blood transfusion from another donor. The volume of blood that will be taken from the patient is expressed in units (1 unit is equivalent to 180 ml of red blood cells). The possibility of donation depends mainly on the patient's blood volume and initial hematocrit. Both indicators determine the endogenous erythrocyte reserve, which can be calculated using the following formula: Endogenous erythrocyte reserve = blood volume [ml] • (hematocrit - 33) : 100 Women: blood volume [ml] = 41 [ml/kg] • body weight [kg ] + 1200 [ml] Men: blood volume [ml] = 44 [ml/kg] • body weight [kg] + 1600 [ml] (for body weight ≥45 kg). Indications for use of Recormon and its single dose are determined according to normograms based on the required volume of donor blood and endogenous erythrocyte reserve. The highest dose should not exceed 1600 IU/kg/week for intravenous administration and 1200 IU/kg/week for subcutaneous administration. Instructions for using the syringe tube:
- Remove one syringe tube from the packaging and make sure that the solution in it is transparent, colorless and does not contain visible impurities. Remove the cap from the syringe.
- Remove one needle from the package, place it on the syringe and remove the protective cap from the needle.
- Remove air from the syringe and needle by holding the syringe vertically and carefully pushing the plunger upward. You should press the plunger until the required dose of Recormon remains in the syringe.
The solution is administered according to generally accepted methods.
Recormon
Treatment of anemia in patients with chronic renal failure: subcutaneously or intravenously for 2 minutes, for hemodialysis patients - through an arteriovenous shunt at the end of the dialysis session. For patients not receiving hemodialysis, it is preferable to administer the drug subcutaneously to avoid accidental entry into peripheral veins. The goal of treatment with the drug is to achieve a hematocrit of 30-35% or eliminate the need for blood transfusion. The weekly increase in hematocrit should not exceed 0.5%. In patients with arterial hypertension, cardiovascular and cerebrovascular diseases, the weekly increase in hematocrit and its weekly indicators should be determined individually depending on the clinical picture. For some patients, the optimal rate is below 30%.
Correction stage: subcutaneous injection, initial dose - 20 IU/kg 3 times a week. In case of insufficient increase in hematocrit (less than 0.5% per week), every 4 weeks the dose can be increased to 40 IU/kg 3 times a week. The total weekly dose can be divided into daily administrations in smaller doses or administered in one dose.
When administered intravenously (slowly over 2 minutes), the initial dose is 40 IU/kg 3 times a week. If there is insufficient increase in hematocrit after 4 weeks, the dose can be increased to 80 IU/kg 3 times a week. If it is necessary to further increase the dose, it can be increased by 20 IU/kg 3 times a week at 4-week intervals. Regardless of the route of administration, the maximum dose should not exceed 720 IU/kg/week.
Maintenance therapy: to maintain the hematocrit at a level of 30-35%, the dose is initially reduced by 2 times from the dose of the previous injection. Subsequently, the maintenance dose is selected for each patient individually, at intervals of 1-2 weeks, so that the hematocrit is maintained at a level of 30-35%. For subcutaneous administration, the weekly dose can be administered in one dose or divided into 3 or 7 administrations per week. When the condition stabilizes, you can switch to a single dose with an interval of 2 weeks; in this case, you may need to increase the dose. Treatment is lifelong and can be interrupted if necessary.
In children, the dose of the drug depends on age: the younger the child, the higher doses of epoetin beta he requires. Since individual response to the drug cannot be predicted, it is advisable to start with the recommended dosage regimen.
Prevention of anemia in premature newborns: subcutaneously, at a dose of 250 IU/kg 3 times a week. The course of treatment is 6 weeks. Treatment should begin as early as possible, preferably from the 3rd day of life.
Prevention and treatment of anemia in cancer patients: for patients with solid tumors receiving chemotherapy with Pt drugs, treatment is indicated if Hb before chemotherapy is not higher than 130 g/l. SC, initial dose - 450 IU/kg/week, divided into 3 or 7 injections. If the increase in Hb is not sufficient, the dose is doubled after 4 weeks. Treatment is continued until 3 weeks after the end of chemotherapy. If during the first cycle of chemotherapy, Hb, despite treatment with the drug, decreases by more than 10 g/l, further use of the drug may be ineffective. An increase in Hb of more than 20 g/l per month or above 140 g/l should be avoided. If Hb increases by more than 20 g/l per month, the dose is reduced by 50%. If Hb exceeds 140 g/L, the drug is discontinued until Hb decreases to 120 g/L or less, and then treatment is resumed at a dose of 50% of the previous weekly dose.
In patients with multiple myeloma and low-grade non-Hodgkin's lymphoma or chronic lymphocytic leukemia, the initial dose of the drug is 450 IU/kg/week. The weekly dose can be divided into 3 or 7 injections. If the increase in Hb is insufficient after 4 weeks (by less than 10 g/l), the dose is doubled. If after 8 weeks of treatment Hb does not increase by at least 10 g/l, the drug should be discontinued. The maximum dose should not exceed 900 IU/kg per week.
For chronic lymphocytic leukemia, treatment should be continued until 4 weeks after the end of chemotherapy. The highest dose should not exceed 900 IU/kg. If within 4 weeks of treatment Hb increases by more than 20 g/l, the dose is reduced by 2 times. If Hb exceeds 140 g/l, treatment must be interrupted until Hb reaches 130 g/l or less, after which therapy is resumed at a dose equal to 50% of the previous initial dose. Treatment should only be restarted if the most likely cause of the anemia is erythropoietin deficiency.
Preparing patients for donated blood collection for subsequent autohemotransfusion: IV or SC 2 times a week for 4 weeks. In cases where the hematocrit (33% or more) allows blood sampling, the drug is administered at the end of the procedure. The dose of the drug is determined individually depending on the patient’s erythrocyte reserve and the volume of blood required for autohemotransfusion. The highest dose should not exceed 1600 IU/kg per week with intravenous administration and 1200 IU/kg/week with subcutaneous administration. Throughout the course of treatment, the hematocrit should not exceed 48%.
Side effects of the drug Recormon
The cardiovascular system. Often: the emergence or intensification of existing hypertension (arterial hypertension) (1%, but ≤10%), especially in the case of a rapid increase in hematocrit; hypertensive crisis with symptoms of encephalopathy (headache and confusion, sensory and motor disorders, speech disorders, walking, tonic-clonic seizures), headache (1%, but ≤10%) (including sudden migraine-like headache), thromboembolic complications in cancer patients (a clear cause-and-effect relationship with taking the drug has not been established). Blood system. A dose-dependent increase in the number of platelets (not beyond the normal range and disappearing with continued therapy), especially after intravenous administration of the drug. Single (≤0.01%): thrombocytosis; Rare (0.01%, but ≤0.1%): thrombosis of shunts (0.01, but ≤0.1%) (possible with inadequate heparinization), especially in patients with a tendency to decrease blood pressure or with complications of an arteriovenous fistula ( for example, stenosis, aneurysm, etc.). Laboratory indicators A decrease in the concentration of ferritin in the blood serum simultaneously with an increase in hematocrit, a decrease in iron metabolism. In patients with uremia - hyperkalemia, hyperphosphatemia. In premature newborns - a decrease in the concentration of ferritin in the blood serum, a slight increase in the number of platelets, especially from the 12th to the 14th day of life. Other Rarely - allergic skin reactions, rash, itching, urticaria, anaphylactoid reactions, reactions at the injection site. Rarely, especially at the beginning of therapy, flu-like symptoms, usually mild or moderate, disappearing after a few hours or a few days: fever, chills, headache, pain in the limbs or bones, general malaise. With subcutaneous administration of erythropoietins, the formation of anti-erythropoietin antibodies with the development of bone marrow red cell aplasia (PRCA) is possible. If PRCA is diagnosed, erythropoietin therapy should be discontinued.
Special instructions for the use of the drug Recormon
Caution must be exercised when prescribing Recormon to patients with refractory anemia in the presence of blast-transformed cells, patients with thrombocytosis, epilepsy and chronic renal failure. Before starting treatment with epoetin beta, it is necessary to exclude deficiency of vitamin B12 and folic acid, as they reduce the effectiveness of Recormon. In some cases, anaphylactoid reactions were noted, so the first dose of the drug must be administered under the supervision of a physician. Acute increases in aluminum levels associated with treatment of renal failure may reduce the effectiveness of epoetin beta. The decision to use Recormon in patients with nephrosclerosis who are not on dialysis must be made individually, since the possibility of a more rapid deterioration of renal function cannot be completely excluded. Patients who have previously been treated with other erythropoietin drugs and who have anti-erythropoietin antibodies and partial red cell aplasia of the bone marrow should not be treated with Recormon until cross-sensitivity to all erythropoietic substances has been determined. In patients with chronic renal failure, a dose-dependent increase in the number of platelets within the normal range is possible, followed by an independent return to normal during treatment with Recormon, especially with intravenous administration of the drug. Therefore, it is necessary to regularly monitor platelet counts during the first 8 weeks of treatment. Newborns may experience a slight increase in platelets, especially between 12 and 14 days of life, so regular monitoring of platelet counts is necessary. Patients who are preparing to donate blood for subsequent autotransfusion may have elevated platelet levels. Therefore, such patients need to undergo weekly platelet monitoring. If they increase by more than 150•109/l compared to the initial value, treatment with Recormon should be stopped. In patients with chronic renal failure, when treated with Recormon, due to increased hematocrit, it is often necessary to increase the dose of heparin during hemodialysis. Early revision of the shunt and timely prevention of thrombosis (for example, taking acetylsalicylic acid) in patients with chronic renal failure are recommended. When treating with Recormon, it is necessary to periodically monitor the level of potassium and phosphate in the blood serum. If hyperkalemia occurs, Recormon should be temporarily discontinued until potassium concentration normalizes. If epoetin beta is prescribed before autologous blood donation, donation guidelines should be followed: blood should only be collected from patients with a hematocrit ≥33% (or a hemoglobin level of at least 110 g/L). Particular caution should be exercised in patients weighing ≤50 kg. The volume of blood for simultaneous sampling should not exceed 12% of the patient’s estimated blood volume. It is recommended to monitor blood pressure, including between dialysis sessions, with a rapid increase in hematocrit levels, and in cancer patients - especially at the beginning of treatment. Increased blood pressure should be corrected with medication, and if there is no effect, a temporary break in treatment should be taken. In most cases, simultaneously with an increase in hematocrit, the concentration of ferritin in the blood serum decreases. Therefore, all patients with anemia of renal origin and with a serum ferritin concentration ≤100 μg/L or transferrin saturation ≤20% are recommended to take oral iron supplements (Fe2+) at a dose of 200–300 mg/day. For premature infants, oral iron therapy at a dose of Fe2+ 2 mg/day should be prescribed as early as possible (no later than the 14th day of life). The dose of iron is adjusted depending on the level of ferritin in the serum. If it remains at a level of ≤100 mcg/ml or there are other signs of iron deficiency, the dose of iron supplements should be increased to 5–10 mg/day and therapy should be continued until signs of iron deficiency resolve. In patients with multiple myeloma, non-Hodgkin's lymphoma or chronic lymphocytic leukemia with a serum ferritin concentration of ≤100 mcg/L or transferrin saturation of ≤25%, intravenous iron supplementation is recommended at a dose of 100 mg/day. Therapy is continued until signs of iron deficiency are eliminated. When using Recormon to increase the volume of autologous blood intended for subsequent autotransfusion, patients need to weigh the benefits of using erythropoietin with the increased risk of thromboembolism when using it in order to avoid blood transfusions. The drug should be prescribed only to patients with moderate anemia (hemoglobin level 10-13 g/dL without iron deficiency) if it is impossible to obtain a sufficient amount of banked blood, and a large amount of blood is required for a planned major surgical intervention. In patients preparing to donate blood for subsequent autotransfusion, with temporary iron deficiency, oral therapy with iron preparations (Fe2+) at a dose of 300 mg/day should be started simultaneously with Recormon therapy and continued until ferritin levels normalize. If, despite oral iron therapy, signs of iron deficiency develop (ferritin level ≤20 mcg/L or transferrin saturation ≤20%), additional IV iron supplementation should be considered. Each syringe tube contains up to 0.3 mg (in dosage forms 1000–5000 IU) or up to 0.6 mg (in dosage form 30,000 IU) phenylalanine. This should be taken into account for patients with severe forms of phenylketonuria. Effect on tumor growth Epoetins are growth factors that primarily stimulate the production of red blood cells. Receptors for erythropoietin can also be expressed on the surface of various tumor cells. Therefore, like other growth factors, epoetins can stimulate the growth of various tumors. Unreasonable deaths were reported in two controlled clinical trials of epoetin in patients with different types of tumors, including head and neck cancer and breast cancer. Patients with cancer also need to regularly monitor platelet levels and hematocrit. Pregnancy and lactation period The results of animal studies demonstrated the absence of teratogenic effects of erythropoietin. There is no sufficient experience with the use of the drug during pregnancy and breastfeeding, therefore epoetin beta should be prescribed only if the possible benefits of its use for the mother outweigh the potential risk for the fetus or infant. Effect on the ability to drive vehicles and operate machinery The drug does not affect the ability to drive vehicles and operate machinery.
Recormon®
Inappropriate use of the drug by healthy people (for example, as doping) can cause a sharp increase in Hb, accompanied by life-threatening complications from the cardiovascular system.
Since anaphylactoid reactions have been reported in isolated cases, the first dose of the drug should be administered under the supervision of a physician.
Platelet, hematocrit and Hb levels should be regularly monitored during Recormon therapy.
Recormon should be used with caution in refractory anemia in the presence of blast-transformed cells, epilepsy, thrombocytosis and chronic liver failure. Before starting treatment with Recormon, it is necessary to exclude deficiency of vitamin B12 and folic acid, because they reduce the effectiveness of therapy.
Iron deficiency should be excluded before starting treatment with Recormon, as well as during the entire period of therapy. If necessary, additional therapy with iron supplements may be prescribed in accordance with clinical recommendations.
When treating patients with severe forms of phenylketonuria, the presence of phenylalanine as an excipient should be taken into account: in each syringe tube - up to 0.3 mg (in dosages of 1000 IU, 2000 IU) or up to 0.6 mg (10,000 IU, 20,000 IU, 30 000 ME), in each cartridge - up to 0.5 mg.
No effect.
The most common causes of incomplete response to treatment with erythropoiesis-stimulating agents are iron deficiency and inflammation (as a result of uremia or advanced metastatic cancer). The following conditions reduce the effectiveness of treatment with drugs that stimulate erythropoiesis: chronic blood loss, bone marrow fibrosis, a sharp increase in aluminum concentration due to hemodialysis, deficiency of folic acid or vitamin B12, hemolysis. If all of the above conditions are excluded and the patient experiences a sudden decrease in Hb levels, reticulocytopenia, and antibodies to erythropoietin are detected, a bone marrow examination must be performed to exclude PRCA. If PRCA develops, Recormon therapy should be discontinued, and patients should not be transferred to therapy with other erythropoiesis stimulants.
PRCA caused by neutralizing anti-erythropoietin antibodies may be associated with therapy with erythropoiesis stimulants, incl. and with Recormon therapy (0.107 cases per 10,000 patient-years when Recormon was used for the treatment of anemia of renal origin intravenously and subcutaneously; 0.158 cases per 10,000 patient-years when Recormon was administered subcutaneously for the treatment of anemia of renal origin). It is not recommended to transfer patients to Recormon therapy if the presence of erythropoietin neutralizing antibodies is suspected or confirmed.
Effect on tumor growth.
Epoetins are growth factors that primarily stimulate the formation of red blood cells. Erythropoietin receptors may be present on the surface of various tumor cells. It cannot be ruled out that drugs that stimulate erythropoiesis can stimulate the growth of any type of malignant tumor.
In clinical studies of the treatment of anemia in cancer patients with epoetin beta, a statistically significant deterioration in survival rate and tumor progression was not recorded.
In patients with chronic kidney disease or cancer receiving chemotherapy,
Episodes of increased blood pressure and worsening of existing arterial hypertension may occur, especially with a sharp increase in Hb content. Increased blood pressure can be eliminated with medication; if there is no effect, a temporary break in treatment with Recormon is necessary. It is recommended to regularly monitor blood pressure (especially at the beginning of therapy), incl. between dialysis sessions in patients with anemia of renal origin. In some patients with chronic kidney disease, a hypertensive crisis with symptoms of encephalopathy may occur even with normal or low blood pressure. Immediate consultation with a physician is necessary, especially if sudden acute migraine-like headaches occur.
During treatment with Recormon, it is recommended to periodically monitor the level of potassium in the blood serum. If hyperkalemia occurs, Recormon should be temporarily discontinued until potassium concentration normalizes.
Patients with chronic kidney disease require an increase in the dose of heparin during a hemodialysis session due to an increase in Hb levels. Occlusion of the dialysis system is possible with inadequate heparinization. Early revision of the shunt and timely prevention of thrombosis (for example, taking acetylsalicylic acid) are recommended.
A moderate dose-dependent increase in the number of platelets within the normal range is possible, especially after intravenous administration of Recormon, followed by an independent return to normal values with continued therapy. In the first 8 weeks of therapy, weekly counts of formed elements and, especially, platelets are necessary.
If Recormon is prescribed before collecting autologous donor blood,
You should follow the recommendations for the donation procedure:
- blood can be taken only from patients with a hematocrit value ≥ 33% (or hemoglobin of at least 110 g/l (6.83 mmol/l));
— special caution should be observed in patients weighing less than 50 kg;
— the volume of blood taken at one time should not exceed 12% of the patient’s estimated blood volume.
It is possible that the platelet count may increase within the normal range in patients receiving Recormon before collecting autologous donor blood, so the platelet count should be monitored weekly. Treatment with Recormon is interrupted if platelets increase by more than 150x109/l or with thrombocytosis.
Treatment with Recormon is indicated only for those patients for whom it is most important to avoid homologous blood transfusion, taking into account the risk-benefit ratio of homologous transfusion.
Possible slight increase in platelet count when preventing anemia in preterm infants
(up to 12-14 days), therefore regular platelet monitoring is recommended.
The decision to use Recormon in patients with nephrosclerosis who are not receiving dialysis must be made individually, because The possibility of a more rapid deterioration of renal function cannot be completely excluded.
In most cases, along with an increase in hemoglobin, the concentration of ferritin in the serum decreases. Therefore, all patients with anemia of renal origin and with a serum ferritin concentration of less than 100 mcg/l or transferrin saturation of less than 20% are recommended to take oral iron supplements (Fe2+) at a dose of 200-300 mg/day.
Patients with oncological and hematological diseases are treated with iron supplements according to the same principles, while patients with multiple myeloma, non-Hodgkin's lymphomas or chronic lymphocytic leukemia with transferrin saturation less than 25% can be administered 100 mg Fe3+ per week intravenously. For premature infants, oral iron therapy at a dose of 2 mg Fe2+ per day should be prescribed as early as possible (at the latest on the 14th day of life). The dose of iron is adjusted depending on the level of serum ferritin. If it persistently remains at a level below 100 mcg/ml or there are other signs of iron deficiency, the dose of iron supplements should be increased to 5-10 mg/day and therapy should be continued until the signs of iron deficiency are relieved.
In patients with moderate anemia before planned major surgery, the drug is prescribed taking into account the benefits of using epoetin beta and the increased risk of thromboembolic complications.
In patients preparing to donate blood for subsequent autotransfusion, since they have indications of temporary iron deficiency, oral therapy with iron preparations (Fe2+) at a dose of 300 mg/day should be started simultaneously with Recormon therapy and continued until ferritin levels normalize. If, despite oral iron replacement therapy, signs of iron deficiency develop (ferritin level ≤ 20 mcg/L or transferrin saturation less than 20%), additional IV iron supplementation should be considered. The Recormon solution in the cartridge contains benzyl alcohol as a preservative, which can cause neurological and other complications in newborns, which can sometimes be fatal.
Impact on the ability to drive vehicles and operate machinery
Studies have not been conducted to study the effect of the drug on the ability to drive vehicles and operate machinery. Based on the mechanism of action and safety profile, Recormon does not have such an effect.
Overdose of the drug Recormon, symptoms and treatment
The therapeutic index of Recormon is very wide. Even at very high concentrations of the drug in the blood serum, no symptoms of intoxication were noted. However, response to epoetin beta varies from person to person and is dose dependent. In case of overdose, hypertension (arterial hypertension) and erythrocytosis may develop. In the case of hypertension (arterial hypertension), it is necessary to exclude overhydration. In the presence of erythrocytosis and hyperhydration, before measures to remove excess fluid from the body, it is necessary to perform a venesection, otherwise a further increase in hematocrit and blood viscosity may occur. For stable hypertension (arterial hypertension), antihypertensive therapy is indicated.