Symptoms
Signs of the disease are different, they depend on where exactly the amyloid deposits are localized, how widespread the disease is, and whether there are complications. Often there is a complex of symptoms reflecting damage to several organs.
With amyloidosis of the gastrointestinal tract the following are observed:
- tongue enlargement;
- difficulty swallowing;
- bowel dysfunction;
- heartburn, nausea;
- stomach ache.
Signs of liver amyloidosis:
- change in liver size;
- pain in the hypochondrium on the right;
- nausea, belching;
- jaundice.
Pancreatic amyloidosis is characterized by dull pain in the left hypochondrium.
Cardiac amyloidosis is expressed in rhythm disturbances, myocardial lesions, and heart failure.
Amyloidosis of the nervous system has the following symptoms:
- peripheral polyneuropathy (numbness of the limbs, tingling, burning sensation);
- headaches, dizziness, increased sweating;
- urinary and fecal incontinence;
- sexual dysfunction.
With amyloidosis of the respiratory system, hoarseness of the voice and bronchitis are observed.
Classification of renal amyloidosis
Systematization of the disease is based on parameters such as etiology and pathogenesis. Based on them, five of its forms are distinguished.
| Form | Characteristics |
| Congenital | AL amyloidosis, the development of which was provoked by the appearance and accumulation of light chains of immunoglobulins in the body tissues. The etiology of the disease still remains unclear, as well as the mechanism of its development. |
| Acquired | AA amyloidosis is secondary, caused by a number of chronic inflammatory processes in which the liver begins to intensively produce the acute phase protein alpha globulin. |
| Family | AF amyloidosis (or "Mediterranean fever") is hereditary. Residents of the Mediterranean (Greeks, Arabs, Armenians) suffer from it. The disease is based on a genetic defect that causes a disruption in the production of fibrillar proteins. |
| Senile | ASC1 amyloidosis develops in elderly patients due to impaired metabolism of serum prealbumin. According to medical statistics, the disease affects about 80% of patients over 80 years of age. |
| Local tumor-like | AE amyloidosis can be triggered by neoplasms of the endocrine system, type 2 diabetes mellitus, and senile dementia of the Alzheimer's type. |
Treatment and prevention
In most cases, amyloidosis is treated at home. If there are complications, the patient may be indicated for hospitalization.
Treatment for amyloidosis includes taking medications and following a number of doctor’s recommendations. But in severe cases, the spleen is removed and kidney or liver transplantation may be required.
The list of medications depends on the location of the deposits, the degree of damage to the body, and existing complications. Thus, with secondary amyloidosis, specific treatment of the primary disease is necessary. In addition, medications are prescribed to eliminate symptoms.
Also, the patient is often prescribed a special diet (restriction of protein and salt intake).
There is no specific preventive program for amyloidosis, since the exact causes of the disease are unknown.
Amyloidosis is a complex disease that requires ongoing treatment. A patient with amyloidosis should be regularly monitored by a specialist and undergo examinations to monitor his health. Medical has modern diagnostic equipment, which allows you to make an accurate diagnosis in the shortest possible time. And the center’s experienced, highly qualified specialists will prescribe effective treatment and ensure proper care for the patient.
Etiology of amyloidosis
The initiating factors for the development of primary amyloid renal dystrophy most often cannot be identified, and it itself often affects not only them, but also the tongue, dermis, thyroid gland, lungs, spleen, heart, liver and intestines.
There are often cases when the disease develops against the background of myeloma, characterized by a malignant course. Secondary renal amyloidosis affects not only the kidneys, but also the blood vessels, lymph nodes, liver and other internal organs. It develops in a number of pathological conditions:
- infectious nature (tuberculosis, malaria);
- purulent etiology (septic endocarditis, bronchiectasis);
- systemic (ankylosing spondylitis, rheumatoid arthritis);
- kidney (neoplastic);
- intestines (colitis, Crohn's disease).
The disease is detected in patients who have undergone extrarenal blood purification by hemodialysis for a long time.
So, what is primary amyloidosis or AL amyloidosis?
Definition of disease.
Amyloid light chain amyloidosis (AL), primary systemic amyloidosis (PSA), or simply primary amyloidosis. The disease occurs when a person's antibody-producing cells (plasmocytes) do not function properly and produce abnormal protein fibers from antibody components called light chains. These light chains form amyloid deposits, which can cause serious damage to various organs. Abnormal light chains in urine are sometimes called "Bence Jones proteins."
Epidemiology.
AL amyloidosis is the most common type of systemic amyloidosis in developed countries, with an estimated incidence of 9 cases per million inhabitants per year. The average age of diagnosed patients is 65 years and less than 10% of patients are under 50 years of age.
Signs and symptoms.
AL amyloidosis can affect a wide range of organs and therefore presents with a range of symptoms. The kidneys are the most commonly affected organ in AL amyloidosis. Symptoms of kidney disease and kidney failure may include fluid retention, swelling, and shortness of breath. Heart complications, which affect more than a third of patients, include heart failure and irregular heartbeat. Other symptoms may include gastrointestinal disturbances, liver enlargement, suppression of adrenal and other endocrine glands, changes in skin color, fatigue and weight loss.
Systemic AL amyloidosis. A. Macroglossia with lateral tongue ridge. B. Bilateral periorbital purpura. C. Pseudo-athletic appearance of secondary diffuse muscle infiltrates. D. Voluminous hepatomegaly due to primary hepatic amyloidosis. E. Diffuse bilateral interstitial lung diseases. F. Enlargement of the submandibular gland. Localized AL amyloidosis. G. Nodular conjunctival amyloidosis. H. Horate amyloid lump.
Diagnosis of the disease.
Diagnosis is based on examination of the area involved showing Congo red-positive amyloid deposits, which stain positive with anti-LC antibody by immunohistochemistry and/or immunofluorescence. Due to the systemic nature of the disease, noninvasive biopsies such as abdominal fat aspiration should be considered before taking biopsies from involved organs to reduce the risk of bleeding.
In addition, blood and urine can be tested for the presence of “light chains,” which can form amyloid deposits, causing disease.
Treatment.
The most effective treatment is autologous bone marrow transplantation using stem cells. However, not all patients are prescribed this type of treatment.
Other treatments may include chemotherapy similar to that used for multiple myeloma. The combination of melphalan and dexamethasone has been found effective in those not suitable for stem cell transplantation, and the combination of bortezomib and dexamethasone is now widespread in clinical use.
Forecast.
Life expectancy with AL amyloidosis depends on the spectrum of organ damage (the main prognostic factor is amyloid heart disease), the severity of damage to individual organs and the hematological response to treatment.
Course of the disease and prognosis
Experts say that the main cause of death in patients is chronic renal failure, which occurs as a result of reactive amyloidosis, which is characterized as an inflammatory infectious process. There is also information that patients with an outbreak of reactive amyloidosis die as a result of an infectious intestinal disease and diarrhea.
Statistics indicate that even with timely detection and treatment of the disease, the average survival rate of patients with renal failure is no more than one and a half years from the date of examination.
Today, leading specialists prolong the lives of patients through bone marrow transplantation, and also perform surgical procedures for kidney transplantation. #diagnosis #urology kidney problems
Morphology and pathogenesis
Amyloid deposition leads to significant concentric thickening of the myocardium, and dilatation of the ventricular cavities is not typical for AS. Amyloid infiltration of the myocardium is accompanied by a deterioration in its mechanical properties and leads to a significant decrease in contractility in combination with severe restrictive disorders of diastolic function. Modern research methods have shown that thickening of the myocardial walls in AS does not always correlate with impaired systolic and diastolic functions. Conventionally, three subtypes of AS can be distinguished: in patients with AL, hereditary ATTR and acquired senile ATTR amyloidosis [16].
In all cases, amyloid is found in the myocardial interstitium in the form of diffuse or nodular deposits. The most common findings are diffuse thickening of the interventricular septum (> 80% of patients) and the posterior wall of the LV. Isolated thickening of the wall of the right ventricle (without thickening of the LV wall) is rare - in 6% of cases, and thickening of the walls of both ventricles - in 40-80% of cases [3, 18]. Thickening of the interatrial septum is observed in 40% of patients. The change in the shape of the LV occurs according to the type of concentric hypertrophy; ventricular dilatation does not develop [19]. The combination of thickening of the myocardial walls in more than half of the patients with low amplitude of the ventricular complex on the electrocardiogram (ECG) in the chest leads (less than 10 mm) and limb leads (less than 5 mm) allows us to confirm the pseudohypertrophic nature of myocardial changes. According to C. Rapezzi et al., the thickness of the myocardial walls in patients with ATTR amyloidosis (especially senile) was significantly greater than in patients with AL amyloidosis [16, 20]. At the same time, patients with AL amyloidosis showed a more significant decrease in the amplitude of the ventricular complex and, accordingly, a lower ratio of QRS amplitude to LV wall thickness. This may indicate that the decrease in QRS amplitude in patients with AL amyloidosis is more related to damage to cardiomyocytes than to interstitial amyloid deposition. It is known that in the pathogenesis of organ damage in patients with AL amyloidosis, the direct toxic effect of free light chains of immunoglobulins on cells is of great importance [16, 18]. In patients with different types of AS, significant vacuolization of cardiomyocytes is detected. Studies in recent years have shown [24] that it is the vacuolization of cardiomyocytes, and not amyloid deposits, that is responsible for the increased echogenicity of the granular type, characteristic of AS and first described by A. Siqueira-Filho et al. [19]. Thus, this ultrasound property of amyloid heart is not specific and cannot be used as a diagnostic criterion.
Damage to cardiomyocytes in patients with AS may be reflected in increased concentrations of troponins and natriuretic peptides. The expression of atrial and medullary nitriuretic peptides has been shown to be increased in ventricular cardiomyocytes from patients with AS, particularly in areas adjacent to amyloid deposits [25]. Although these substances are not specific markers of amyloid cardiac damage, they are considered highly sensitive indicators and, therefore, an important prognostic factor in systemic amyloidosis [26].
Amyloid can be deposited in the area of the heart valves, often causing their thickening, noticeable on an echocardiogram [18, 27]. Thickening of the papillary muscles has been described [28], but valve function is often preserved.
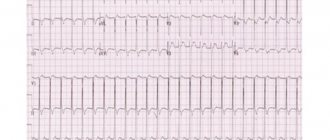
Amyloid can damage the conduction system of the heart; cases of amyloid masses penetrating directly into the sinoatrial node have been described. Conduction disorders are most often represented by incomplete blockade of the anterior branch of the left bundle branch (20%), complete blockade of the right (4–19%) or left bundle branch (2–7%), and first degree atrioventricular block (18–33%) [16, 18]. Conduction disturbances, as a rule, occur in senile AS, which may be due to the longer course of this variant of the disease [3]. Amyloid deposition in the area of adrenergic synapses disrupts the neurohumoral regulation of heart function [29], which may be one of the risk factors for tachyarrhythmias. In 5–27% of patients, atrial fibrillation is detected; more rare rhythm disturbances are ventricular tachycardia and junctional rhythm. Some changes in the ECG occur in almost all (> 90%) patients with AS.
A distinctive property of AL amyloidosis is its deposition in the walls of the coronary arteries [15], which can cause myocardial ischemia [30]. Some patients complain of angina pectoris [31], although changes in the angiogram may be absent [32]. Myocardial infarction may develop. Pathological Q-waves and changes in repolarization without clinical signs of myocardial infarction or indication of it in the anamnesis are found in more than half of the patients, which allows these changes to be interpreted as pseudo-infarction. It appears that mimicking infarct changes is associated with nodular deposition of electrically inactive amyloid, which creates a scar effect. In the later stages of the disease, pericardial effusion is detected in almost half of patients [18].
Quantitative assessment of the total mass of amyloid deposits in the heart is possible using SAP scintigraphy. SAP is a normal plasma glycoprotein whose levels increase dramatically in amyloid deposits of any type as a result of reversible calcium-dependent binding to the amyloid fibril. After intravenous administration, the radiotracer labeled with SAP is distributed between the circulating and amyloid-bound SAP pools in proportion to their volume. In this way, images can be obtained for qualitative and quantitative assessment of amyloid deposits. The method is especially useful in monitoring the course of amyloidosis, including for the purpose of assessing the effectiveness of treatment.
Diagnosis of amyloid kidney dystrophy
During the latent stage, diagnosis of the disease is seriously difficult, so laboratory diagnostics of urine and blood are performed. The first make it possible to detect protein and leukocytes, the level of which is constantly increasing, as well as an admixture of blood and bodies from coagulated protein, blood cells, and kidney epithelium. Blood tests detect decreased albumin levels, increased globulin levels, abnormally elevated lipid and/or lipoprotein levels, and electrolyte imbalances.
In the presence of pathological processes, various diagnostic techniques make it possible to identify their specific manifestations. So:
- the electrocardiogram registers arrhythmia;
- echocardiography reveals myocardial pathologies, characterized by structural changes in the heart muscle;
- ultrasound diagnostics of the peritoneum determines an increase in the volume of internal organs such as the liver and spleen;
- X-ray examination of the gastrointestinal tract determines the deterioration of contraction of the stomach walls, too slow or, on the contrary, rapid movement of barium through the intestines;
- Ultrasound scanning of the kidneys reveals their significant increase;
A biopsy of the kidney, rectal mucosa and liver is required.