Simponi®
Infections
Before prescribing Simponi®, during therapy and for 5 months after its completion, patients should be closely monitored for the development of infections. If severe infections or sepsis develop, therapy should be discontinued.
Simponi® should not be administered to patients with clinically significant active infection. Caution should be exercised when using Simponi® in patients with a history of chronic infection or recurrent infection. Patients are advised to avoid exposure to potential risk factors for infection whenever possible.
Patients receiving therapy with TNF inhibitors are at greater risk of developing an infectious process. There are reports of the development of bacterial (including sepsis and pneumonia), mycobacterial (tuberculosis), invasive fungal and opportunistic infections, incl. with a fatal outcome in patients receiving TNF inhibitors, including the drug Simponi®. In some cases, serious infections have developed in patients receiving concomitant immunosuppressant therapy, which, like the disease itself, predisposes to the development of infections. Patients with new cases of infectious diseases should be carefully assessed. The use of Simponi® should be discontinued in cases of severe infections or sepsis and appropriate antibacterial or antifungal therapy should be prescribed until the infectious process is controlled.
Before initiating treatment with Simponi® in patients who have lived in or visited areas where invasive mycoses are endemic, such as histoplasmosis, coccidioidomycosis or blastomycosis, the possible benefits and risks of treatment with Simponi® should be carefully weighed.
Tuberculosis
Cases of tuberculosis development have been reported in patients receiving therapy with Simponi®. In most cases these were extrapulmonary or disseminated forms of tuberculosis.
Before starting treatment with Simponi®, it is necessary to evaluate risk factors for tuberculosis (including close contact with patients with active tuberculosis) and exclude the presence of latent or active tuberculosis. Treatment of latent tuberculosis should be carried out before using Simponi®. In case of active tuberculosis, therapy with Simponi® is contraindicated.
In patients with a history of latent tuberculosis or active tuberculosis, for which adequate treatment cannot be confirmed, anti-tuberculosis therapy should be administered before prescribing Simponi®.
Tests for latent tuberculosis may be falsely negative, especially in patients who are immunocompromised or have severe disease. Before prescribing Simponi®, it is necessary to consider the possibility of treating latent tuberculosis if patients have relevant risk factors, despite negative test results for latent tuberculosis. The decision to prescribe anti-tuberculosis therapy in such patients should be made only after consulting a TB specialist, weighing the risk of having latent tuberculosis and the risk of using anti-tuberculosis therapy.
Patients receiving Simponi® should be monitored for signs and symptoms of active tuberculosis during and after treatment, including patients who have tested negative for latent tuberculosis.
All patients should be informed about symptoms suspicious for tuberculosis (prolonged cough, weight loss, low-grade fever) and the need to seek medical help if they occur. If a tuberculosis infection develops during therapy with Simponi®, the use of the drug should be discontinued and anti-tuberculosis therapy prescribed
Hepatitis B virus reactivation
As with treatment with other immunosuppressants, therapy with TNF-α inhibitors was accompanied by reactivation of the hepatitis B virus in chronic carriers of the virus (with a positive test for surface antigen), including the development of death. All patients should be examined to exclude viral hepatitis B before starting therapy. Chronic carriers of the hepatitis B virus should be closely monitored before starting treatment, during treatment and for several months after stopping treatment with Simponi®.
Data on the effectiveness of the combined use of antiviral therapy and TNF-α inhibitors in patients who are chronic virus carriers are not available.
If the viral infection reactivates, treatment with Simponi® should be discontinued and appropriate antiviral therapy should be prescribed.
Malignant tumors
The possible role of therapy with TNFα inhibitors in the development of malignant tumors has not been established, however, based on current data, the risk of developing lymphomas, leukemia and other malignant tumors during anti-TNFα therapy cannot be excluded. Caution should be exercised when prescribing TNF inhibitors to patients with a history of malignancy or when continuing therapy if a malignancy develops.
Malignant tumors in children
During post-marketing studies, cases of malignancy, some fatal, have been reported among children, adolescents and young adults (<22 years of age) receiving TNF inhibitors (initiation of therapy ≤18 years of age). Lymphoma was reported in approximately half of the cases. Other cases include a range of different malignancies, including malignancies not typically seen in children and adolescents. Most patients received concomitant therapy with immunosuppressants such as methotrexate, azathioprine, or 6-mercaptopurine. The role of TNF inhibitors in the development of malignant tumors in children and adolescents remains unclear.
Lymphoma
In controlled clinical trials with all TNF inhibitors, including Simponi®, cases of lymphoma were reported more often in patients treated with TNF inhibitors than in the control group. In clinical trials of Simponi® phase 2 and 3, the incidence of lymphoma in patients receiving Simponi® was higher than the expected frequency in the general population. Patients with rheumatoid arthritis and other chronic inflammatory diseases, especially those with high disease activity and/or patients undergoing long-term immunosuppressant therapy, are at several times higher risk of developing lymphoma than in the general population, even without treatment with TNF inhibitors.
Leukemia
Cases of acute and chronic leukemia have been reported during post-marketing use of TNF inhibitors for the treatment of rheumatoid arthritis and other diseases. Even in the absence of TNF inhibitor therapy, patients with rheumatoid arthritis may be at increased risk (approximately 2-fold) of developing leukemia compared with the general population.
Other malignant tumors
In controlled phase 2 and 3 clinical trials of Simponi® in patients with rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis, the incidence of other malignancies (excluding non-melanoma skin cancer) was similar in the Simponi® group and the control group.
In a clinical study evaluating the use of Simponi® in patients with severe persistent asthma, malignant tumors were more common in patients receiving Simponi® than in the control group. The significance of this fact has not been established.
In a clinical trial using another TNFα inhibitor, infliximab, in patients with moderate to severe COPD, a higher incidence of lung, head and neck tumors was reported in the TNF inhibitor group compared with controls. Therefore, caution should be exercised when prescribing TNF inhibitors to patients with COPD, as well as to high-risk patients, such as chronic smokers.
Congestive heart failure
During treatment with TNF inhibitors, cases of increase or development of congestive heart failure have been observed, incl. during therapy with Simponi®. In clinical studies using other TNF inhibitors, progression of heart failure and increased mortality due to heart failure were observed. The effect of Simponi® has not been studied in patients with congestive heart failure. Simponi® should be used with caution in patients with mild heart failure (NYHA class I-II). Patients should be monitored and if new or worsening signs of heart failure occur, therapy with Simponi® should be discontinued.
Neurological disorders
The use of TNF inhibitors, including the drug Simponi®, in rare cases was accompanied by the appearance or increase in clinical and/or radiological signs of demyelinating diseases of the central nervous system (including multiple sclerosis) and the peripheral nervous system. In patients with existing or newly emerging demyelinating diseases, the benefits and risks of treatment with TNF inhibitors should be carefully weighed before prescribing Simponi®. If such diseases develop, therapy with Simponi® should be discontinued.
Surgery
Data on the safety of the use of the drug Simponi® in patients receiving surgical treatment, incl. arthroplasty are limited. When planning operations, it is necessary to take into account the long T1/2. When performing operations, patients receiving therapy with Simponi® require careful monitoring of infections and their timely treatment if they occur.
Immunosuppression
There is a potential impact of TNF inhibitors, including Simponi®, on immunity against infections and tumors due to the blockade of inflammation and modulation of cellular responses mediated through TNFα.
Autoimmune processes
A relative deficiency of TNFα during therapy with TNF inhibitors can lead to the development of autoimmune processes. If clinical symptoms of lupus-like syndrome appear and tests for antibodies to double-stranded DNA are positive, therapy with Simponi® should be discontinued.
Combined use of the drug Simponi® and the drug anakinra
The combined use of anakinra and another TNF inhibitor, etanercept, was associated with the development of serious infections and neutropenia in clinical studies and did not lead to additional clinical benefit. Due to the nature of the adverse reactions observed with this combination therapy, similar toxicities may occur with combination therapy of anakinra and other TNF inhibitors. In this regard, the combined use of Simponi® and anakinra is not recommended.
Combined use of Simponi® and abatacept
In clinical studies, the combined use of TNF inhibitors and abatacept was associated with an increased risk of infections, including serious infections, compared with the use of TNF inhibitors alone, without increased clinical effect. Due to the nature of the adverse reactions observed with combination therapy of TNF inhibitors and abatacept, the combination of Simponi® and abatacept is not recommended.
Change of biological base anti-inflammatory drugs
When switching from one biological product to another, patients should continue to be monitored for signs of infection.
Hematological reactions
During post-marketing experience, there are reports of pancytopenia, leukopenia, neutropenia and thrombocytopenia in patients receiving TNF inhibitors, including Simponi®. Caution should be exercised when treating patients with cytopenia or a history of severe cases of cytopenia with Simponi®.
Patients who develop signs of hematologic abnormalities (persistent fever, bruising, bleeding, pallor) should be evaluated immediately. In case of severe hematological disorders, Simponi® therapy should be discontinued.
Allergic reactions
In a post-marketing study of Simponi®, serious systemic hypersensitivity reactions, including anaphylactic reactions, were described. Some cases are described after the first administration of the drug. In cases of anaphylactic reactions or other serious allergic reactions, the use of Simponi® should be discontinued and appropriate therapy should be prescribed.
Vaccination
Patients receiving Simponi® may receive concomitant vaccination, but not live vaccines. There are no data on vaccination response, risk of infection or transmission of infection with live vaccines in patients receiving Simponi®. Patients with psoriatic arthritis treated with Simponi® in one phase 3 study showed an effective B-cell immune response to the pneumococcal polysaccharide vaccine. A comparable number of psoriatic arthritis patients treated and not treated with Simponi® experienced at least a twofold increase in antibody titers. The proportion of patients responding to pneumococcal vaccine in the Simponi® group and the control group of patients receiving methotrexate was lower than in patients not receiving methotrexate. Overall, the data obtained indicate that Simponi® did not suppress the humoral immune response to this vaccine.
Sensitivity to latex
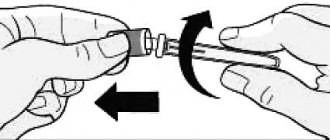
The needle guard on the prefilled syringe is made of dry natural rubber that contains latex, which may cause allergic reactions in latex-sensitive patients.
Elderly patients
In phase 3 studies in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, the incidence of adverse events, serious adverse events, and serious infections with Simponi did not differ between patients aged 65 years and older and younger patients. The number of patients over 65 years of age with ulcerative colitis was not sufficient to establish whether their response to therapy differed from that of patients with ulcerative colitis aged 18 to 65 years. In general, a higher incidence of infections has been noted in older patients, so caution should be used when treating older patients.
Kidney and liver failure
Special studies of the drug Simponi® in patients with impaired renal and liver function have not been conducted. Caution must be exercised when treating patients with impaired liver function.
Clinical effectiveness in the treatment of rheumatoid arthritis
The efficacy and safety of Simponi® were studied in three multicenter, randomized, double-blind, placebo-controlled studies in more than 1,500 patients aged ≥18 years with moderate to severe active rheumatoid arthritis, diagnosed according to the American College of Rheumatology (ACR) criteria. at least 3 months before screening. Simponi® was administered subcutaneously in doses of 50 mg or 100 mg (with or without MTX) every 4 weeks. The duration of the placebo-controlled phase was 24 weeks.
In general, the clinical effectiveness of Simponi® in doses of 50 mg and 100 mg did not differ significantly.
In all phase 3 studies, the proportion of patients adequately responding to treatment at 14 and 24 weeks was higher in the Simponi® drug groups than in the control groups. The response was observed 4 weeks after the start of treatment with Simponi® (when results were first assessed) and was maintained for 24 weeks.
When treated with Simponi® compared to placebo, a clinically and statistically significant improvement in quality of life was detected. There was a statistically significant improvement in work ability and a decrease in fatigue.
Clinical efficacy in the treatment of psoriatic arthritis
The safety and efficacy of Simponi® were studied in a multicenter, randomized, double-blind, placebo-controlled study in 405 adult patients with active psoriatic arthritis who had not responded to NSAID or disease-modifying anti-inflammatory drugs. In this study, the time from diagnosis of psoriatic arthritis was at least 6 months before inclusion in the study, the diameter of the plaque skin lesion was at least 2 cm. Patients with various subtypes of psoriatic arthritis were included in the study, incl. polyarthritis without rheumatoid nodules (43%), asymmetric peripheral arthritis (30%), arthritis of the distal interphalangeal joints (15%), spondylitis with peripheral arthritis (11%) and mutilating arthritis (1%). Previous TNF antagonist therapy was an exclusion criterion. Simponi® was administered subcutaneously in doses of 50 mg or 100 mg every 4 weeks with or without methotrexate. Randomized patients were assigned to placebo (n=113), Simponi® 50 mg (n=146) and Simponi® 100 mg (n=146). The primary endpoint was the proportion of patients achieving 20% improvement in ACR criteria (ACR 20) at 14 weeks. Treatment results were compared with placebo over 24 weeks.
In general, the clinical effectiveness of Simponi® in doses of 50 mg and 100 mg did not differ significantly.
Improvement in the main parameters characterizing disease activity was noted already at the first examination (4 weeks) after the start of treatment and persisted for 24 weeks. Response rates at 14 weeks were similar in patients with different subtypes of psoriatic arthritis, incl. polyarthritis without rheumatoid nodules, asymmetric peripheral arthritis, arthritis of the distal interphalangeal joints and spondylitis with peripheral arthritis. The number of patients with arthritis mutilans was insufficient to evaluate effectiveness. The response rate to treatment with Simponi® was comparable in patients receiving and not receiving methotrexate.
When treated with Simponi®, an improvement in activity indicators of psoriatic arthritis was observed, incl. number of swollen joints, number of painful joints, dactylitis and enthesitis. In addition, patients treated with Simponi® showed significant improvement in skin and nail psoriasis.
Treatment with Simponi® resulted in significant improvements in physical function as well as quality of life. Work capacity has increased significantly, and the time spent with the patient by those providing care or treatment has been reduced.
Clinical effectiveness in the treatment of ankylosing spondylitis
The safety and effectiveness of Simponi® were studied in a multicenter, randomized, placebo-controlled study in 356 adult patients with active ankylosing spondylitis. The study included patients who had persistent signs of active ankylosing spondylitis despite treatment with NSAIDs or disease-modifying anti-inflammatory drugs. They had not previously received TNF antagonists. Patients with complete ankylosis of the spine were excluded from the study. The drug Simponi® was administered subcutaneously every 4 weeks. Randomized patients were assigned to placebo (n=78), Simponi® 50 mg (n=138) and Simponi® 100 mg (n=140). The primary endpoint was the proportion of patients achieving ASAS 20 improvement at 14 weeks. Efficacy was compared with placebo over 24 weeks.
In general, the clinical effectiveness of Simponi® in doses of 50 mg and 100 mg did not differ significantly.
Compared with placebo, treatment with Simponi® resulted in a significant reduction in symptoms at 14 and 24 weeks. Improvement in the main indicators of ankylosing spondylitis activity was noted at the first assessment of treatment results (4 weeks) and was maintained for 24 weeks.
Treatment with Simponi® caused a significant increase in physical function, which was assessed after 14 and 24 weeks. In patients treated with Simponi®, improvements in physical function were maintained for 24 weeks. Quality of life also improved significantly at 14 and 24 weeks. In addition, significant improvements in sleep and work ability were found.
Clinical efficacy in the treatment of ulcerative colitis
The effectiveness of Simponi® was studied in two randomized, placebo-controlled, double-blind studies in adult patients.
The Remission Induction Study (PURSUIT-Induction) evaluated patients with moderate to severe active ulcerative colitis (Mayo score 6 to 12, endoscopy subscale score 2 or greater) who had insufficient or no response to standard therapy or who had dependence on corticosteroids. For the dose-finding portion of the study, patients were randomized into 4 groups: 400 mg Simponi® SC at week 0 and 200 mg at week 2 (400/200 mg), 200 mg Simponi® SC at weeks 0 and 100 mg in week 2 (200/100 mg), 100 mg of Simponi® SC in week 0 and 50 mg in week 2 (100/50 mg), placebo SC in week 0 and week 2 (placebo). In the dose confirmation portion of the study, 771 patients were randomized into 3 groups: 400 mg Simponi® SC at week 0 and 200 mg at week 2, 200 mg Simponi® SC at week 0 and 100 mg at week 2, subcutaneous placebo at week 0 and week 2. Concomitant administration of continuous doses of aminosalicylates, corticosteroids, and/or oral immunosuppressants was allowed. This study assessed the effectiveness of Simponi® over a 6-week period.
The results of the PURSUIT-Maintenance study are based on the evaluation of 463 patients who achieved a clinical response to previous induction therapy with Simponi. Patients were randomized into 3 groups: 50 mg of Simponi®, 100 mg of Simponi®, or placebo; the drug was administered subcutaneously every 4 weeks. Concomitant administration of continuous doses of aminosalicylates, corticosteroids, and/or oral immunosuppressants was allowed. This study assessed the effectiveness of Simponi® over a period of 54 weeks.
Table. Main results of the PURSUIT-Induction and PURSUIT-Maintenance studies
| PURSUIT-Induction | ||
| Placebo n=256 | Simponi® 200/100 mg n=257 | |
| Percentage of patients | ||
| Patients with clinical response at 6 weeksa | 30% | 52%b |
| Patients in clinical remission at week 6 | 6% | 19%b |
| Patients with mucosal healing at 6 weeks | 29% | 43%b |
| Change in baseline IBDQ score at week 6 | 15±31 | 27±34b |
| PURSUIT-Maintenance | ||
| Placebo n=156 | Simponi®100 mg n=154 | |
| Percentage of patients | ||
| Maintenance of response (patients with clinical response at 54 weeks)e | 31% | 51%b |
| Persistent remission (patients in clinical remission at 30 and 54 weeks)f | 15% | 29%g |
| Persistent mucosal healing (patients with mucosal healing at 30 and 54 weeks) | 27% | 44%b |
n is the number of patients; a is defined as a decrease in the initial Mayo score by ≥30% and by ≥3 points, accompanied by a decrease in the score on the rectal bleeding scale by ≥1, or if the score on this scale is from 0 to 1; bp≤0.001, for the golimumab group compared with placebo; c was defined as a Mayo scale score ≤2 points, in the absence of results on individual subscales >1; d is defined as an endoscopy subscale score of 0 to 1; The activity of ulcerative colitis in patients was assessed by partial Mayo score every 4 weeks (loss of response was confirmed by endoscopy). Therefore, patients who maintained response were in the stage of continuous clinical response at each assessment over 54 weeks; f patient in remission at both week 30 and week 54 (with no evidence of loss of response at any time point during week 54) to achieve sustained remission; g p = 0.003, for the golimumab group compared with placebo.
Immunogenicity
Antibodies to golimumab were detected in 5% (105/2115) of patients with rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis treated with Simponi® for 52 weeks in phase 3 studies. The frequency of antibody formation was comparable in patients with various rheumatic diseases. In patients receiving concomitant therapy with methotrexate, the frequency of antibodies was lower than in patients who took Simponi® without methotrexate (3% (41/1262) and 8% (64/853), respectively).
Antibodies to golimumab were detected in 3% (26/946) of patients with ulcerative colitis treated with Simponi® for 54 weeks in phase 2 and 3 studies. In 68% (21/31) of antibody-positive patients, neutralizing antibodies were detected in vitro.
When treated with concomitant immunosuppressive therapy (azathioprine, 6-mercaptopurine and methotrexate), the number of patients with antibodies to golimumab (2%; 6/389) was lower than when treated without concomitant therapy (4%; 28/809).
The presence of antibodies to golimumab may increase the risk of injection site reactions. The low incidence of antibodies to golimumab does not allow definitive conclusions about the relationship between immunogenicity and clinical efficacy or safety.
Since the immunogenicity assay is product specific and specific to each individual reagent kit, comparisons of antibody levels to different products are not valid.
Use in pediatrics
Specific studies of Simponi® have not been conducted in children. The drug is contraindicated in children and adolescents under 18 years of age, because safety and effectiveness in this category of patients have not been studied.
Impact on the ability to drive vehicles and other mechanisms
The drug Simponi® may have a slight effect on the ability to drive a car and operate machinery due to the possible development of adverse reactions from the nervous system and the organ of vision.
Simponi 50mg/0.5ml 0.5ml/№1
Infections
Before prescribing Simponi, during therapy and for 5 months after its completion, patients should be closely monitored for the development of infections. If severe infections or sepsis develop, therapy should be discontinued.
Simponi should not be administered to patients with clinically significant active infection. Caution should be exercised when using Simponi in patients with a history of chronic infection or recurrent infection. Patients are advised to avoid exposure to potential risk factors for infection whenever possible.
Patients receiving TNF inhibitor therapy are at greater risk of developing an infectious process. There are reports of the development of bacterial (including sepsis and pneumonia), mycobacterial (tuberculosis), invasive fungal and opportunistic infections, incl. with a fatal outcome in patients receiving TNF inhibitors, including the drug Simponi. In some cases, serious infections have developed in patients receiving concomitant immunosuppressant therapy, which, like the disease itself, predisposes to the development of infections. Patients with new cases of infectious diseases should be carefully assessed. The use of Simponi should be discontinued in cases of severe infections or sepsis and appropriate antibacterial or antifungal therapy should be prescribed until the infection is controlled.
Before initiating treatment with Simponi in patients who have lived in or visited areas where invasive mycoses are endemic, such as histoplasmosis, coccidioidomycosis or blastomycosis, the possible benefits and risks of treatment with Simponi should be carefully weighed.
Tuberculosis
Cases of tuberculosis development have been reported in patients receiving therapy with Simponi. In most cases these were extrapulmonary or disseminated forms of tuberculosis.
Before starting treatment with Simponi, the patient must be carefully examined to identify both active and latent tuberculosis. The examination should include a thorough history taking, incl. it is necessary to exclude a history of tuberculosis and contact with patients with tuberculosis, and also to clarify whether immunosuppressant therapy is currently or has been carried out previously. The necessary screening tests (chest x-ray, tuberculin test) should be performed. It should be taken into account that in seriously ill patients and in patients with immunosuppression, tests for latent tuberculosis may be falsely negative.
If active tuberculosis is diagnosed, therapy with Simponi cannot be started.
If latent tuberculosis is suspected, a phthisiatrician should be consulted.
In all cases described below, the possible risks and expected benefits of therapy with Simponi should be carefully assessed.
In patients with latent tuberculosis, it is necessary to undergo anti-tuberculosis therapy before prescribing Simponi.
In patients with multiple or significant risk factors for developing tuberculosis, but in whom latent tuberculosis is not confirmed by testing, the need for anti-tuberculosis therapy should be considered before initiating therapy with Simponi.
The need for anti-tuberculosis therapy should be considered before initiating treatment with Simponi in patients with a history of active or latent tuberculosis for whom an adequate course of therapy cannot be confirmed.
Cases of the development of active tuberculosis have been reported in patients receiving therapy with Simponi during and after treatment for latent tuberculosis. Patients receiving Simponi should be monitored for signs and symptoms of active tuberculosis, incl. in patients with negative test results for latent tuberculosis, patients receiving therapy for latent tuberculosis, and patients with a history of treatment for tuberculosis.
Patients should be informed about symptoms suspicious for tuberculosis (prolonged cough, weight loss, low-grade fever) and the need to seek medical help if they occur during or after therapy with Simponi.
Hepatitis B virus reactivation
As with treatment with other immunosuppressants, therapy with TNF-α inhibitors was accompanied by reactivation of the hepatitis B virus in chronic carriers of the virus (with a positive test for surface antigen), incl. with the development of death. All patients should be examined to exclude viral hepatitis B before starting therapy. Chronic carriers of the hepatitis B virus should be closely monitored before starting treatment, during treatment and for several months after stopping treatment with Simponi.
Data on the effectiveness of the combined use of antiviral therapy and TNF-α inhibitors in patients who are chronic virus carriers are not available.
If the viral infection reactivates, treatment with Simponi should be discontinued and appropriate antiviral therapy should be prescribed.
Malignant tumors
The possible role of therapy with TNFα inhibitors in the development of malignant tumors has not been established, however, based on current data, the risk of developing lymphomas, leukemia and other malignant tumors during anti-TNFα therapy cannot be excluded. Caution should be exercised when prescribing TNF inhibitors to patients with a history of malignancy or when continuing therapy if a malignancy develops.
Malignant tumors in children
During post-marketing studies, cases of malignancies, some fatal, have been reported among children, adolescents and young adults (<22 years of age) receiving TNF inhibitors (initiation of therapy ≤18 years of age). Lymphoma was reported in approximately half of the cases. Other cases include a range of different malignancies, including malignancies not typically seen in children and adolescents. Most patients received concomitant therapy with immunosuppressants such as methotrexate, azathioprine, or 6-mercaptopurine. The role of TNF inhibitors in the development of malignant tumors in children and adolescents remains unclear.
Lymphoma and leukemia
In controlled clinical trials with all TNF inhibitors, including Simponi, cases of lymphoma were reported more often in patients treated with TNF inhibitors than in the control group. In phase 2b and phase 3 clinical studies, the incidence of lymphoma in patients receiving Simponi was higher than the expected incidence in the general population. Cases of leukemia have been reported during post-marketing use of TNF inhibitors. Because The risk of developing lymphoma and leukemia is increased in patients with rheumatoid arthritis with long-standing, highly active inflammatory disease, and risk assessment is difficult.
In the post-registration period, rare cases of the development of heptolienal T-cell lymphoma have been reported during therapy with TNF inhibitors. This rare type of T-cell lymphoma is very aggressive and usually fatal. Almost all cases have been reported in patients with Crohn's disease or in patients with ulcerative colitis. Most cases have been reported to occur in adolescent or young adult males. Almost all of these patients were treated with azathioprine or 6-mercaptopurine with a TNF inhibitor at or before diagnosis. The possible risk of concomitant use of azathioprine or 6-mercaptopurine and Simponi should be carefully assessed. The risk of developing hepatolienal lymphoma in patients receiving treatment with TNF inhibitors cannot be excluded.
Patients with rheumatoid arthritis and other chronic inflammatory diseases, especially those with high disease activity and/or patients undergoing long-term immunosuppressant therapy, are at several times higher risk of developing lymphoma than in the general population, even without treatment with TNF inhibitors.
Non-lymphocytic malignant tumors
In controlled phase 2b and phase 3 clinical trials of Simponi in patients with rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and ulcerative colitis, the incidence of other malignancies (excluding nonmelanoma skin cancer) was similar in the Simponi group and the control group.
In clinical
Simponi, 50 mg/0.5 ml, solution for subcutaneous administration, 0.5 ml, 1 pc.
Live vaccines and medicinal products containing infectious agents
Patients taking Simponi® may receive concomitant vaccination, but not with live vaccines (see section “Interaction with other drugs”). There are insufficient data on vaccine response or the potential for secondary transmission of infection with live vaccines in patients receiving TNF inhibitor therapy. The use of live vaccines may result in clinical infections, including disseminated infection. The use of drugs containing infectious agents such as live attenuated bacteria (eg, BCG bladder instillations for the treatment of cancer) may lead to clinical infections, including disseminated infection. The simultaneous use of drugs containing infectious agents and the drug Simponi® is not recommended.
Allergic reactions
Sensitivity to latex
The protective cap of the needle of the disposable syringe and disposable syringe pen is made of dry natural rubber containing latex, which may cause allergic reactions in patients sensitive to latex.
Special patient groups
Children
Vaccination
If possible, it is recommended to complete routine vaccinations in accordance with the vaccination schedule before starting therapy with Simponi® in pediatric patients.
Elderly patients
In phase 3 studies in patients with RA, PsA, AS and UC, the incidence of adverse events, serious adverse events and serious infections when treated with Simponi® did not differ between patients aged 65 years and older and younger patients. However, when treating elderly patients, care should be taken and special attention should be paid to the occurrence of infections. Patients aged 45 years or older were excluded from the non-radiographic axial spondyloarthritis study.
Kidney and liver failure
Special studies of the drug Simponi® in patients with impaired renal and hepatic function have not been conducted. Caution must be exercised when treating patients with impaired liver function.
Possibility of medical errors
The drug Simponi® is registered in two dosages: 50 mg/0.5 ml and 100 mg/1.0 ml in the form of a solution for subcutaneous administration. It is very important to use the correct dosage in accordance with the section "Dosage and Administration". Caution should be exercised during use to avoid underdosing or overdosing.
Clinical effectiveness in the treatment of rheumatoid arthritis
The efficacy and safety of Simponi® were studied in three multicenter, randomized, double-blind, placebo-controlled studies in more than 1,500 patients aged ≥18 years with moderate to severe active RA. diagnosed according to American College of Rheumatology (ACR) criteria at least 3 months before screening: in adult patients who have had an inadequate response to DMARD therapy, including MTX; in adult patients who have not previously received MTX and TNF inhibitors: in patients who have previously received one or more TNF inhibitors. Simponi® was administered subcutaneously in doses of 50 mg or 100 mg (with or without MTX) every 4 weeks. The duration of the placebo-controlled phase was 24 weeks.
In general, the clinical effectiveness of Simponi® in doses of 50 mg and 100 mg did not differ significantly.
In all phase 3 studies, the proportion of patients adequately responding to treatment at 14 and 24 weeks was higher in the Simponi® drug groups than in the control groups. Response according to ACR criteria was observed 4 weeks after the start of treatment with Simponi® (when results were first assessed) and was maintained over the subsequent 24 and 104 weeks.
Treatment with Simponi® compared with placebo showed clinically and statistically significant improvements in physical function and quality of life. There was a statistically significant improvement in work ability and a decrease in fatigue.
In a study of adult patients who had not previously received MT, the vdH-S scale, which takes into account the number and severity of joint erosions and narrowing of the joint spaces of the hands and feet, was used to evaluate the effect of Simponi® on radiographic progression. The number of patients without new erosions or with changes in the initial vdH-S scale score ≤0 was significantly higher in patients in the Simponi® group than in the control groups (p = 0.003). The effect on radiographic progression achieved at week 52 was maintained until week 104.
In patients who remained in the study and continued therapy with Simponi®, the effect on radiological parameters persisted from 104 to 256 weeks.
Clinical efficacy in the treatment of psoriatic arthritis
The safety and efficacy of Simponi® were studied in a multicenter, randomized, double-blind, placebo-controlled study in 405 adult patients with active PsA. those who have not responded to therapy with non-steroidal anti-inflammatory drugs or DMARDs. In this study, the time from diagnosis of PsA was at least 6 months before enrollment, and the diameter of the skin plaque lesion was at least 2 cm. Patients with different subtypes of PsA were included in the study. including polyarthritis without rheumatoid nodules (43%), asymmetric peripheral arthritis (30%), arthritis of the distal interphalangeal joints (15%), spondylitis with peripheral arthritis (11%) and mutilating arthritis (1%). Previous TNF antagonist therapy was an exclusion criterion. The drug Simponi® was administered subcutaneously in doses of 50 mg or 100 mg every 4 weeks with or without MT. Randomized patients were assigned to placebo (n = 113), Simponi® 50 mg (n = 146), and Simponi® 100 mg (n = 146). The primary endpoint was the proportion of patients achieving 20% improvement according to ACR criteria (ACR 20) at 14 weeks. Treatment results were compared with placebo over 24 weeks.
In general, the clinical effectiveness of Simponi® in doses of 50 mg and 100 mg did not differ significantly.
Improvement in the main parameters characterizing disease activity was noted already at the first examination (4 weeks) after the start of treatment and persisted for 24 weeks. Response rates at 14 weeks were similar in patients with different PsA subtypes. including polyarthritis without rheumatoid nodules. asymmetric peripheral arthritis, arthritis of the distal interphalangeal joints and spondylitis and peripheral arthritis. The number of patients with arthritis mutilans was insufficient to evaluate effectiveness. The response rate to treatment with Simponi® was comparable in patients receiving and not receiving MTX.
When treated with Simponi®, improvements were observed in indicators of psoriatic arthritis activity, including the number of swollen joints, the number of painful joints, dactylitis and enthesitis. In addition, patients treated with Simponi® showed significant improvement in skin and nail psoriasis.
Treatment with Simponi® resulted in significant improvements in physical function as well as quality of life. Work capacity has increased significantly, and the time spent with the patient by those providing care or treatment has been reduced.
In patients who remained in the study and continued therapy with Simponi®, the effect on clinical parameters persisted from weeks 104 to 256.
Clinical effectiveness in the treatment of axial spondyloarthritis
Clinical effectiveness in the treatment of ankylosing spondylitis
The safety and effectiveness of Simponi® were studied in a multicenter, randomized, placebo-controlled study in 356 adult patients with active AS. The study included patients who had persistent signs of AS activity despite treatment with nonsteroidal anti-inflammatory drugs or DMARDs and who had not previously received TNF antagonists. Patients with complete ankylosis of the spine were excluded from the study. Simponi® was administered subcutaneously every 4 weeks. Randomized patients were assigned to placebo (n = 78), Simponi® 50 mg (n = 138), and Simponi® 100 mg (n = 140). The primary endpoint was the proportion of patients achieving ASAS 20 improvement at 14 weeks. Efficacy was compared with placebo over 24 weeks.
In general, the clinical effectiveness of Simponi® in doses of 50 mg and 100 mg did not differ significantly.
Compared with placebo, treatment with Simponi® resulted in a significant reduction in symptoms at 14 and 24 weeks. Improvement in the main indicators of AS activity was noted at the first assessment of treatment results (4 weeks) and persisted for 24 weeks.
Treatment with Simponi® contributed to a significant improvement in physical function, which was assessed after 14 and 24 weeks. In patients treated with Simponi®, improvements in physical function were maintained for 24 weeks. Quality of life also improved significantly after just 14 weeks. In addition, significant improvements in sleep and work ability were found.
In patients who remained in the study and continued therapy with Simponi® d, the effect on clinical parameters persisted from weeks 24 to 256.
Clinical effectiveness in the treatment of non-radiographic axial siondyloarthritis.
The safety and effectiveness of Simponi® was studied in a multicenter, randomized study. double blind. A placebo-controlled study (GO-AHEAD) in 197 adult patients with severe active non-radiographic axial spondyloarthritis (patients meeting ASAS criteria for axial spondyloarthritis but not meeting criteria for the modified New York AS classification). Patients were included in the study if they still had signs of disease activity (AS Disease Activity Index (BASDAI) ≥4 and Visual Analog Scale (VAS) criteria for total back pain ≥4, each on a scale of 0 to 10 cm), despite treatment with NSAIDs, and who have not previously received therapy with other biological drugs, including therapy with TNF antagonists. Patients were randomly assigned to placebo or Simponi® 50 mg subcutaneously every 4 weeks. From week 16, the open-label phase of the study began, in which all patients received therapy with Simponi® 50 mg subcutaneously every 4 weeks until week 48, assessing efficacy for 52 weeks and safety for 60 weeks. Approximately 93% of patients who received Simponi® at the beginning of the open-label phase of the study (week 16) continued to receive treatment until the end of the study (week 52).
Analyzes were performed on two endpoints for all patients (AT, N=197) and patients with objective signs of inflammation (OSI, N=158, such as increased CRP concentrations and/or evidence of sacroiliitis on MRI at baseline). This study assessed the effectiveness of Simponi® over 16 weeks compared to placebo. The primary endpoint was the proportion of patients achieving ASAS 20 improvement at week 16. The main results are presented in Table 2 and described below.
Table 2. Key findings from the GO-AHEAD study at week 16.
| Improvements in signs and symptoms | ||||
| All patients (AT) | Patients with objective signs of inflammation (OSI) | |||
| Placebo | Simponi® 50 mg | Placebo | Simponi® 50 mg | |
| pa | 100 | 97 | 80 | 78 |
| Patients who responded to treatment, % | ||||
| ASAS 20 | 40% | 71%** | 38% | 77%** |
| ASAS 40 | 23% | 57%** | 23% | 60%** |
| ASAS 5/6 | 23% | 54%** | 23% | 63%** |
| ASAS partial remission | 18% | 33%* | 19% | 35%* |
| ASDAS-Cb <1.3 | 13% | 33%* | 16% | 35%* |
| BASDAI 50 | 30% | 58%** | 29% | 59%** |
| Inhibition of inflammation in the sacroiliac joints, determined using MRI | ||||
| Placebo | Simponi® 50 mg | Placebo | Simponi® 50 mg | |
| nс | 87 | 74 | 69 | 61 |
| Mean change in sacroiliac joints as determined by MRI using the SPARCCd score | -0,9 | -5,3** | -1,2 | -6,4** |
an is the number of patients randomized and receiving assigned therapy;
b concentration of CRP on the AS Disease Activity Scale (ASDAS-C), (AT-placebo, N=90; AG-Simponi® 50 mg, N=88; OSI-placebo, N=71; OSI-Simponi® 50 mg, N= 71);
cn is the number of patients for whom there are initial and MRI data obtained at week 16;
d SPARCC (Spondyloarthritis Research Consortium of Canada);
** p<0.0001 for Sim pony1 compared to placebo;
* p<0.05 for Sim pony compared to placebo.
A statistically significant improvement in symptoms and signs of severe active non-radiographic axial spondyloarthritis was shown in patients treated with Simponi® 50 mg compared to patients treated with placebo at week 16 (see Table 2). An improvement in the main parameters characterizing the activity of the disease was noted already at the first examination (4 weeks) after the start of treatment with Simponi®.
A statistically significant reduction in sacroiliac joint inflammation, as measured by SPARCC MRI, was observed in patients treated with Simponi 50 mg compared to patients treated with placebo at week 16 (see Table 2). Pain, assessed as total back pain and night pain by VAS, and disease activity, measured by ASDAS-C, also showed a statistically significant improvement at week 16 compared with baseline values in patients treated with Simponi® 50 mg, compared with patients receiving placebo at week 16 (p < 0.0001).
There was a statistically significant improvement in spinal mobility, as measured by the AS Metrological Index (BASMI), and a statistically significant improvement in physical function, as measured by the AS Functional Index (BASFI), in patients treated with Simponi® 50 mg, compared with patients treated with placebo (p<0.0001). Patients treated with Simponi® had significantly greater improvements in quality of life as assessed by the ASQoL and EQ-5D questionnaires, physical and mental components by the SF-36 questionnaire, as well as a significant improvement in productivity as assessed by the WPAI questionnaire, compared with patients who received placebo.
Patients with objective evidence of inflammation (OSI) also demonstrated statistically significant results for all endpoints described previously. The improvements observed at week 16 among patients receiving Simponi 50 mg therapy were maintained in patients remaining in the study through week 52.
Clinical efficacy in the treatment of ulcerative colitis
The effectiveness of Simponi® was studied in two randomized, placebo-controlled, double-blind studies in adult patients.
The Remission Induction Study (PURSUIT-Induction) evaluated patients with moderate to severe active UC (Mayo score 6 to 12, endoscopy subscale score 2 or greater) who had poor or no response to standard therapy or who were dependent. from corticosteroids. In the dose confirmation portion of the study, 761 patients were randomized to 3 groups: 400 mg Simponi® subcutaneously at week 0 and 200 mg at week 2, 200 mg Simponi® subcutaneously at week 0 and 100 mg at week 2, placebo subcutaneously at week 0 and at week 2. Simultaneous administration of constant doses of aminosalicylates was allowed. oral corticosteroids and/or immunosuppressants. This study assessed the effectiveness of Simponi® over a 6-week period.
The results of the PURSUIT-Maintenance study are based on the evaluation of 456 patients who achieved a clinical response to previous therapy with Simponi® during the induction phase. Patients were randomized into 3 groups: 50 mg of Simponi®, 100 mg of Simponi®, or placebo; the drug was administered subcutaneously every 4 weeks. Simultaneous administration of constant doses of aminosalicylags was allowed. oral corticosteroids and/or immunosuppressants. This study assessed the effectiveness of Simponi® over a period of 54 weeks. Patients who completed 54 weeks of treatment in the maintenance study continued treatment in the extension phase of the clinical study, with efficacy assessed at week 216.
Table 3. Main results of the PURSUIT-Induction and PURSUIT-Maintenance studies.
| PURSUIT-Induction | |||
| Placebo N=251 | Simponi® 200/100 mg, N=253 | ||
| Percentage of patients | |||
| Patients with clinical response at 6 weeksa | 30% | 51%** | |
| Patients in clinical remission at 6 weeksb | 6% | 18%** | |
| Patients with mucosal healing at 6 weeksc | 29% | 42%* | |
| PURSUIT-Maintenance | |||
| Placebod N=154 | Simponi® 50 mg N=151 | Simponi® 100 mg N=151 | |
| Percentage of patients | |||
| Maintenance of response (patients with clinical response at week 54)e | 31% | 47%* | 50%** |
| Persistent remission (patients in clinical remission at 30 and 54 weeks)f | 16% | 23%g | 28%* |
Where:
N number of patients;
** p ≤0.001;
* p ≤0.01;
adefined as a decrease in the initial Mayo score by ≥30% and by ≥3 points, accompanied by a decrease in the score on the rectal bleeding scale by ≥1 or if the score on this scale is from 0 to 1;
b was defined as a Mayo scale result ≤2 points in the absence of results on individual subscales >1;
c is defined as an endoscopy subscale score of 0 to 1;
d induction stage only;
e patients' ulcerative colitis activity was assessed by partial Mayo score every 4 weeks (loss of response confirmed by endoscopy). Therefore, patients who maintained response were in the stage of continuous clinical response at each assessment over 54 weeks;
f the patient must be in remission at both week 30 and week 54 (with no evidence of loss of response at any time point during week 54) to achieve sustained remission;
g among patients weighing less than 80 kg, more patients receiving golimumab 50 mg maintenance therapy demonstrated sustained clinical remission compared with patients receiving placebo.
The number of patients who had sustained mucosal healing (patients with mucosal healing at 30 and 54 weeks) was greater in the Simponi® group (42% in the 50 mg group (nominal p value <0.05) and 42% in the 100 mg (nominal p value <0.005)) compared with the placebo group (27%).
Among the 54% of patients (247/456) receiving concomitant corticosteroid therapy at the start of maintenance therapy, the proportion of patients with corticosteroid-free clinical response at week 54 was greater in the Simponi 50 mg groups (38%, 30/ 78) and at a dose of 100 mg (30%, 25/82), compared with the placebo group (21%, 18/87). The number of patients who discontinued corticosteroids by week 54 was greater in the Simponi 50 mg (41%, 32/78) and 100 mg (33%, 27/82) groups compared with placebo (22 %, 19/87). Among patients recruited into the extension phase of the clinical trial, the proportion of patients who did not require corticosteroid therapy was largely maintained over 216 weeks.
By week 6, the use of Simponi® significantly improved the quality of life of patients, as evidenced by an improvement in disease-specific indicators (IBDQ) compared to baseline. In patients receiving maintenance therapy with Simponi®, improvement in quality of life, as measured by the IBDQ, was maintained over 54 weeks.
Approximately 63% of patients who received Simponi therapy at the beginning of the extension phase of the clinical study (week 56) continued to receive therapy until the end of the study (last dose of golimumab at week 212).
Immunogenicity
Antibodies to golimumab were detected by enzyme-linked immunosorbent assay (ELISA) in 5% (105/2062) of patients with RA, PsA and AS who received Simponi® for 52 weeks in phase 3 studies. The frequency of antibody formation was comparable in patients with various rheumatic diseases. In patients receiving concomitant MTX therapy, the incidence of antibody formation was lower than in patients who took Simponi® without MTX (3% [41/1235] and 8% [64/827], respectively).
Antibodies to golimumab were detected by ELISA in 4% (4/93) of patients with non-radiographic axial spondyloarthritis who received Simponi® for 16 weeks.
Antibodies to golimumab were detected by ELISA in 3% (26/946) of patients with UC who received Simponi® for 54 weeks in phase 2 and 3 studies. In 68% (21/31) of antibody-positive patients, neutralizing antibodies were detected in vitro. When treated with concomitant immunosuppressive therapy (azathioprine, 6-mercaptopurine and methotrexate), the number of patients with antibodies to golimumab (1%; 4/308) was lower than when treated without concomitant therapy (3%; 22/638).
To detect antibodies to golimumab in the pJIA study, an ELISA method was used to detect total antibodies, including those associated with the drug. Due to the higher sensitivity and lower error of the method, a higher frequency of detection of antibodies to golimumab was expected compared to the previous ELISA method. In a phase 3 study of pJIA up to week 48, ELISA testing for total antibodies, including drug-related antibodies, detected antibodies to golimumab in 40% (69/172) of children receiving golimumab; Most of them have an antibody titer below 1:1000. An effect on the serum concentration of golimumab was observed at a titer >1:100, an effect on efficacy was noted only when a titer reached >1:1000; a small number of children with a titer >1:1000 were reported (N = 8). Among children with antibodies to golimumab, neutralizing antibodies were detected in 39% (25/65). Since cases with low antibody titres were most often reported, increasing the frequency of antibody detection by ELISA with the determination of total antibodies, including those associated with the drug, has no apparent effect on drug concentration, efficacy and safety. Thus, no new information on the safety profile has been identified. The presence of antibodies to golimumab may increase the risk of injection site reactions. The low incidence of antibodies to golimumab does not allow definitive conclusions about the relationship between immunogenicity and clinical efficacy or safety.
Since the immunogenicity assay is product specific and specific to each individual reagent kit, comparisons of antibody concentrations to different products are not valid.
Clinical efficacy in the treatment of polyarticular juvenile idiopathic arthritis
The efficacy and safety of Simponi® was studied in a randomized, double-blind, placebo-controlled, randomized withdrawal study (GO KIDS). which included 173 patients (age range from 2 to 17 years) with active pJIA, with at least 5 joints with active inflammation and an inadequate response to MTX therapy. The study included children with polyargicular JIA (polyarthritis with positive or negative rheumatoid factor, widespread oligoargitis, juvenile psoriatic arthritis or systemic JIA without systemic symptoms at the time of examination). The median number of joints with active inflammation at baseline was 12; the median CRP concentration was 0.17 mg/dL.
Part 1 of the clinical trial was a 16-week open-label phase in which patients received Simponi® at a dose of 30 mg/m2 (maximum 50 mg) subcutaneously every 4 weeks along with MTX. 154 patients who achieved a therapeutic response to ACR Ped 30 at week 16 were included in Part 2 of the clinical trial, which consisted of a randomized withdrawal phase, where patients received Simponi® 30 mg/m2 (maximum 50 mg) + MTX, or placebo + MTX every 4 weeks. After an exacerbation of the disease, patients received Simponi® 30 mg/m2 (maximum 50 mg) + MTX. At week 48, patients were enrolled in the long-term extension phase of the clinical trial.
Therapeutic response of ACR Ped 30, 50, 70 and 90 was observed as early as week 4 of this clinical study.
At week 16, 87% of patients had ACR Ped 30 response, 79%, 66%, 36% of patients had ACR Ped 50, ACR Ped 70, and ACR Ped 90 response, respectively. At week 16, 34% of patients had inactive disease, defined by the presence of all of the following: absence of joints with active arthritis, absence of fever, rash, serositis, and splenomegaly. hepatomegaly and generalized lymphadenopathy associated with JIA, absence of active uveitis, normal erythrocyte sedimentation rate (<20mm/hour) and CRP concentration (<1.0 mg/dl), physician assessment of disease activity <5 mm on the VAS scale; duration of morning stiffness <15 minutes.
At week 16, clinically significant improvements relative to baseline were achieved for all ACR Ped levels (Table 4).
Table 4. Positive dynamics of ACR Ped components in week 16a.
| Median percentage of positive dynamics | ||
| Simponi® 30 mg/m2 nb = 173 | ||
| General assessment of the disease by a physician (VASc 0–10 cm) | 88% | |
| Overall assessment of general well-being by the patient or his parents (VAS 0–10 cm) | 67% | |
| Number of joints with active inflammation | 92% | |
| Number of joints with limited range of motion | 80% | |
| Physical function according to the CHAQd questionnaire | 50% | |
| ESR (mm/hour)e | 33% | |
a baseline = week 0;
b “n”—number of included patients;
c VAS - Visual Analogue Scale;
d CHAQ (Child Health Assessment Questionnaire) - Questionnaire for assessing the health status of children;
e ESR (mm/hour) - erythrocyte sedimentation rate (millimeters per hour).
The primary endpoint of the percentage of patients with an ACR Ped 30 response at week 16 who did not experience a flare from week 16 to week 48 was not met. The majority of patients did not experience a sudden exacerbation of the disease in the period from week 16 to week 48 (59% in the Simponi® + MTX group, 53% in the placebo + MTX group; p = 0.41).
A subgroup analysis of the primary endpoint stratified by baseline CRP concentration (≥1 mg/dL versus <1 mg/dL) showed a greater incidence of acute disease exacerbation in the placebo + MTX group compared with the golimumab + MTX group among patients with Baseline CRP ≥1 mg/dL (87% vs. 40%; p=0.0068).
At week 48, there was a therapeutic response to ACR Ped 30 in 53% and 55% of patients in the Simponi® + MTX and placebo + MTX groups, respectively; the state of inactive disease was achieved in 40% and 28% of patients in the Simponi® + MT and placebo + MT groups, respectively.
Impact on the ability to drive vehicles and other mechanisms
Simponi® may have a slight effect on the ability to drive a car and use machinery. After administration of Simponi®, dizziness may develop. In this case, you should not drive a car or operate machinery.
Golimumabum
Infectious and parasitic diseases:
upper respiratory tract infections (rhinitis, nasopharyngitis, pharyngitis, laryngitis), bacterial infections (cellulitis), viral infections (influenza and herpes), bronchitis, sinusitis, superficial fungal infections, septic shock, sepsis, tuberculosis, lower respiratory tract infections respiratory tract (pneumonia), opportunistic infections (invasive fungal infections (histoplasmosis, coccidioidomycosis, pneumocystosis), bacterial, atypical mycobacterial and protozoal), abscess, bacterial arthritis, reactivation of hepatitis B, pyelonephritis, infectious bursitis.
Benign, malignant tumors and unspecified neoplasms:
skin cancer, squamous cell carcinoma and myelocytic nevus, lymphoma, leukemia.
From the hematopoietic system:
anemia, leukopenia, thrombocytopenia, pancytopenia, aplastic anemia.
From the immune system:
allergic reactions (bronchospasm, hypersensitivity, urticaria), positive reaction to autoantibodies, anaphylactic reactions, systemic vasculitis, sarcoidosis.
From the endocrine system:
thyroid disorders (hypothyroiditis, hyperthyroiditis and goiter).
From the side of metabolism:
increased blood glucose and increased lipids.
From the nervous system:
dizziness, paresthesia, headache, depression, insomnia, demyelinating diseases (central and peripheral forms), balance disorders, personality disorders.
From the organs of vision:
blurred vision (blurred vision), conjunctivitis, allergic reactions (redness, irritation).
From the cardiovascular system:
arterial hypertension, congestive heart failure (newly detected or worsening of existing), arrhythmia, angina pectoris, thrombosis (including deep vein and arterial thrombosis), Raynaud's disease, hyperemia.
From the respiratory system:
bronchial asthma and accompanying symptoms (shortness of breath, bronchial hyperactivity), interstitial lung disease.
From the digestive system:
constipation, dyspepsia, pain in the gastrointestinal tract, nausea, inflammatory diseases of the gastrointestinal tract (gastritis, colitis), reflux, stomatitis, increased activity of liver transaminases, cholelithiasis, liver dysfunction.
For the skin and subcutaneous tissues:
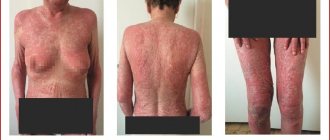
itching, rash, psoriasis (newly detected or worsening of an existing disease, palmoplantar, pustular), urticaria, systemic vasculitis, alopecia, dermatitis.
From the musculoskeletal system and connective tissue:
lupus-like syndrome, bone fractures.
From the urinary system:
diseases of the bladder, kidneys.
From the genital organs and mammary glands:
diseases of the mammary glands, menstruation disorders.
General violations:
hyperthermia, asthenia, chest discomfort.
Local reactions:
erythema, urticaria, induration, pain, bruising, itching, irritation, paresthesia at the injection site, slow healing at the injection site.