Author:
Amelicheva Alena Aleksandrovna medical editor
Quick Transition Treatment of Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia (AIHA) is a group of rare acquired hematological diseases and syndromes characterized by hemolysis (destruction) of red blood cells due to the formation of autoantibodies to the antigens of these blood components.
AIHA can occur at any age; women are more likely to be affected (60%). Pathology develops gradually or suddenly.
There are two main types of AIHA: heat (autoantibodies are most active/attack red blood cells at a temperature of 37-40 ° C) and cold - with cold hemolysins (autoantibodies are most active at temperatures less than 30 ° C, red blood cells are destroyed even with local exposure to cold, for example, when a person drinks cold water or washes his hands in cold water).
The prevalence of AIHA is 1 case per 70-80 thousand people per year.
Causes
The causes of the disease are not well understood. Today it is known that approximately 50% of cases of t-AIHA are idiopathic (develop spontaneously). Whereas x-AIHA are associated with other diseases or occur simultaneously with them: autoimmune, oncological, infectious (systemic lupus erythematosus, lymphocytic leukemia, non-Hodgkin lymphoma, Epstein-Barr virus, cytomegalovirus, mycoplasma pneumonia, hepatitis, HIV). Taking certain medications, for example, penicillin drugs (drug-induced AIHA), can also play a role in the development of AIHA.
3. Symptoms and diagnosis
Autoimmune lysis of red blood cells can begin suddenly and end just as suddenly, without any intervention. In other cases, the disease develops gradually and remains asymptomatic for a long time.
There is also a steadily progressive course, for example, with the patient’s increased sensitivity to cold.
The most typical symptoms of autoimmune hemolytic anemia are “unreasonable” (actually caused by tissue hypoxia) fatigue, weakness, palpitations, shortness of breath, an uncomfortable feeling of heaviness in the left hypochondrium (splenomegaly occurs, the predominant destruction of red blood cells occurs in the spleen).
Sometimes there is pain in the heart and/or lower back, nausea and frequent vomiting. With more severe pathology, jaundice is observed. Some pathogenetically different types of the disease lead to “black urine syndrome”; in the clinic of others, an angiospastic response to cold is formed and secondary Raynaud’s syndrome, cold urticaria or, in the most severe disorders of tissue trophism in the extremities, acrogangrene develops.
Suspicion of hemolytic anemia arises, as a rule, in the presence of an appropriate clinical picture and complaints, as well as a characteristic medical history (risk factors, see above). However, the final diagnosis is established and confirmed by a thorough, most often multiple laboratory blood test (direct and indirect Coombs tests, general clinical, biochemical analysis, etc.).
About our clinic Chistye Prudy metro station Medintercom page!
Symptoms
In some people, the disease may be asymptomatic, especially if AIHA develops gradually (the destruction of red blood cells is not as extensive). Basically, the symptoms of AIHA are similar to the clinical manifestations of other types of anemia (weakness, increased fatigue, pale skin).
Symptoms of more severe disease (rapid destruction of red blood cells) include jaundice of the skin and visible mucous membranes, weakness, fatigue with tachycardia and shortness of breath on exertion, abdominal discomfort, a feeling of fullness, cold extremities and peripheral cyanosis (with cold AIHA) . Physical examination reveals splenomegaly (pathological enlargement of the spleen) of varying degrees.
If AIHA develops against the background of another pathology, symptoms of the underlying disease may predominate, for example: enlarged and painful lymph nodes, fever, severe back and leg pain, headaches, vomiting, diarrhea, dark brown urine color.
1.General information
Anemia (anemia, literally Greek “exsanguination”) is a syndrome that accompanies many diseases and pathological conditions. Anemia is a decrease in the concentration of hemoglobin in the blood, which is most often associated with a deficiency of its carriers, red blood cells - erythrocytes. By combining, due to the high iron content in its chemical composition, with pulmonary oxygen and then distributing it bound to the bloodstream, hemoglobin ensures oxygenation of all tissues and organs. Accordingly, a decrease in the specific number of red blood cells and/or a lack of hemoglobin in them, which is the essence of anemia, leads to disturbances in cellular respiration and the development of a polymorphic clinical picture. The variety of symptoms is determined mainly by the specific etiopathogenetic basis of anemia as a syndrome: it can be vitamin deficiency, unbalanced nutrition, diseases of the hematopoietic system (in particular, bone marrow), massive simultaneous blood loss or chronic bleeding, infections, parasitosis, etc.
Hemolytic anemia occurs much less frequently than other anemias (iron deficiency, hematopoietic, etc.) and, in turn, represents a large group of congenital and acquired diseases, the general essence of which is the abnormally rapid destruction of red blood cells - “hemolytic” means “caused by dissolution, disintegration blood." In the heterogeneous group of such anemias, the most common are autoimmune hemolytic anemias, which again include several variants.
With regard to autoimmune hemolytic anemia, not all questions have yet been clarified, and this disease is being studied today as intensively as its relative rarity allows. It has been established, in particular, that manifestation can occur at any age, that females predominate among those affected, and that the types of course and outcome options vary widely. Most researchers are currently inclined to the hypothesis of the polyetiological (multi-cause) nature of the disease.
A must read! Help with treatment and hospitalization!
Treatment of autoimmune hemolytic anemia
The first line of treatment for warm autoimmune hemolytic anemia is glucocorticosteroids. High-dose prednisolone monotherapy can be prescribed for 3-6 weeks with a gradual dose reduction or discontinuation thereafter. However, due to the severe side effects of such treatment, short-term pulse therapy with methylprednisolone or dexamethasone is increasingly used. Approximately one third of patients achieve remission, while the rest require maintenance therapy with GCS.
If steroid therapy is ineffective, the possibility of splenectomy (surgical removal of the spleen), biological therapy (rituximab) or immunosuppressive therapy (cyclosporine, azathioprine, etc.) is considered.
The prognosis depends on the root cause of the disease, timely initiation and proper treatment. A favorable prognosis (achievement of remission, long-term remission) is entirely associated with a positive therapeutic response in the patient and the absence of complications.
Patients with cold autoimmune hemolytic anemia are advised to avoid triggers (cold, including infusions of cold solutions), and also undergo treatment for the underlying disease (for example, lymphoma), if the development of c-AIHA is associated with it. First-line therapy for x-AIHA is rituximab. GCS is not the treatment of choice due to the low therapeutic response in patients. Splenectomy is also ineffective. In severe cases, plasmapheresis is recommended.
Features and advantages of treatment of autoimmune hemolytic anemia at the Rassvet clinic
Autoimmune hemolytic anemia is a fairly rare blood disease; as it is studied, the criteria for diagnosis and treatment are refined and updated.
Hematologists at the Rassvet Clinic adhere to standardized diagnostic criteria and therapeutic approaches developed by the International Consensus Group on AIHA. We recognize the importance of an accurate diagnostic search in these patients, since the course of the disease and effective treatment depend on the type of antibodies involved. To diagnose primary AIHA, we use a monospecific direct antiglobulin test; we make sure to find out the reasons for the manifestation of secondary AIHA, i.e., we identify the underlying disease with which the development of hemolytic anemia is associated.
When treating t-AIHA, we use glucocorticosteroids. Rituximab is prescribed only in the early stages of severe disease, as well as in the absence of a rapid therapeutic response to steroids. Rituximab in combination with an antineoplastic drug is prescribed to patients with c-AIHA in cases where their condition requires treatment based on clinical signs.
Hematology of Dawn is represented by highly qualified doctors who have extensive experience in identifying and treating difficult to diagnose and rare diseases. In their work, our specialists use international treatment protocols and use only safe, proven and effective methods.
Anemia, pathology of hemostasis, oncohematology
Materials are presented from the RUDN textbook
Anemia. Clinic, diagnosis and treatment / Stuklov N.I., Alpidovsky V.K., Ogurtsov P.P. – M.: Medical Information Agency LLC, 2013. – 264 p.
Copying and reproducing materials without indicating the authors is prohibited and is punishable by law.
Acquired hemolytic anemia, in which increased destruction of red blood cells is the result of the destructive effects of autoantibodies directed against unchanged membrane antigens of the patient's own red blood cells, are called autoimmune.
It is assumed that, as with other autoimmune diseases, the reasons for the production of anti-erythrocyte autoantibodies lie in a dysfunction of the body’s immune system, in particular, in a violation of the ability of suppressor T cells to keep under control autoimmune or “forbidden” clones of B lymphocytes capable of producing antibodies against antigens of one's own body.
The frequency of AIHA is 1:80,000 in the population; women are more often affected.
The course and prognosis of AIHA is largely determined by the type of autoantierythrocyte antibodies circulating in the patient’s blood.
Autoimmune antibodies often have a structure of IgG, less often - IgM and IgA. They can exhibit the greatest activity at high body temperature (warm antibodies), or at low temperature (cold antibodies), or fix on red blood cells at low temperatures, and have a damaging effect at body temperature (biphasic Donath-Landsteiner antibodies).
According to the mechanism of their damaging action, anti-erythrocyte antibodies are divided into:
- agglutinins, which cause sticking (agglutination) of red blood cells;
- hemolysins, which cause the destruction (lysis) of red blood cells with the participation of the activated complement system;
- opsonins that promote phagocytosis of red blood cells.
Depending on the serological characteristics, there are two main types of agglutinins: complete and incomplete.
Complete agglutinins cause red blood cells to stick together in any medium: water-salt or colloidal. Most complete agglutinins are IgM. Due to the large size of the molecule, autoantibodies of the IgM type are able to overcome the negative electrostatic interaction between erythrocytes, therefore, even in a saline environment, serum containing complete IgM agglutinins causes the erythrocytes to stick together if there are antigens on their surface against which these IgM autoantibodies are directed.
Incomplete antibodies often have an IgG or IgA structure and are unable to cause agglutination of red blood cells in a water-salt environment. The gluing of erythrocytes by incomplete antibodies can only occur in a colloidal medium, in cases where they manage to change the electrostatic forces that cause the repulsion of erythrocytes. In patients with autoimmune hemolytic anemia caused by incomplete agglutinins, antibodies are fixed on the surface of red blood cells to the corresponding antigen (usually the Rh system), causing their sensitization, but not the process of intravascular adhesion (agglutination).
Hemolysins are less common than agglutinins. Depending on the temperature optimum, heat, cold and two-phase hemolysins are distinguished. Most hemolysins are of the IgG type, less often of the IgM and IgA type. By fixing on the surface of red blood cells that have the corresponding antigen against which antibodies are directed, hemolysins cause destruction of red blood cells, mainly through the activation of the complement system, the components of which have a proteolytic effect.
Opsonins are antibodies that promote the phagocytosis of red blood cells by monocytes and macrophages. Opsonins are usually found simultaneously with cold hemolysins. Opsonins are detected by erythrophagocytosis, which occurs after incubation of the test blood with the patient’s serum.
AIHA is classified into distinct variants depending on the serological features or type of autoantibodies that cause its development. In addition, each serological variant distinguishes idiopathic and symptomatic forms that occur in patients who already have other diseases.
AIHA classification
I. AIHA with incomplete thermal agglutinins:
- idiopathic;
- symptomatic in patients with lymphoproliferative diseases (chronic lymphocytic leukemia, lymphomas), SLE, RA, periarteritis nodosa, ovarian tumors, thyroid diseases.
II. AIHA with thermal hemolysins:
- idiopathic;
- symptomatic in patients with myelofibrosis and chronic lymphocytic leukemia.
III. AIHA with complete cold agglutinins:
- idiopathic;
- symptomatic in patients after viral pneumonia, infectious mononucleosis, in patients with lymphoproliferative diseases, Waldenström's macroglobulinemia, monoclonal gammopathy and in patients with chronic hepatitis.
IV. AIHA with biphasic cold hemolysins of the Donath-Landsteiner type:
- idiopathic;
- symptomatic in patients with advanced forms of syphilis, with viral infections (cytomegalovirus).
AIHA with incomplete thermal agglutinins
AIHA with incomplete thermal agglutinins is the most common form (80–85%) of this type of hemolytic anemia. AIHA with incomplete thermal agglutinins (ITA) occurs in all age groups, but more often in middle-aged people, with a frequency of 1:80,000 of the population. Among the patients, there is a slight predominance of women (55-60%). Idiopathic and symptomatic forms occur with equal frequency.
Clinic
AIHA with NTA, as a rule, begins gradually and is characterized by slowly progressive pallor, jaundice of the skin and mucous membranes, and sometimes low-grade fever. Due to the patient's gradual adaptation to the slow decrease in hemoglobin levels, the patient's general condition suffers slightly. Less common is the acute and subacute onset of the disease with a decrease in hematocrit by 5% or more every 24 hours, while jaundice quickly increases, sudden general weakness, cyanosis, shortness of breath, tachycardia and other signs of cardiovascular failure appear. Subsequently, the disease, as a rule, acquires a chronic course, in which episodes of increased hemolysis are replaced by a state of clinical and hematological compensation. Relapses of hemolysis can be provoked by infections, surgical interventions and pregnancy.
A slight enlargement of the liver is observed in 1/2 - 1/3 of patients, and a moderate enlargement of the spleen is observed in more than 1/2 of patients.
The mechanism of hemolysis in AIHA with NTA is intracellular in nature, i.e. the destruction of red blood cells is carried out by macrophages that have receptors for Ig. Since autoantibodies have their own Ig structure, macrophages phagocytose those red blood cells on the surface of which antibodies are fixed, most often of the IgG type. Whole red blood cells (erythrophagocytosis) or their fragments can undergo phagocytosis. In the latter case, red blood cells decrease in size and take the form of microcytes.
The most intense intracellular hemolysis occurs in the spleen, where a physiological slowdown in blood flow occurs in the sinuses and, consequently, the duration of contact of sensitized erythrocytes and macrophages lining the walls of the sinuses is prolonged.
A similar type of intracellular hemolysis in AIHA with NTA also occurs in the liver, bone marrow and other organs rich in macrophages.
Laboratory data
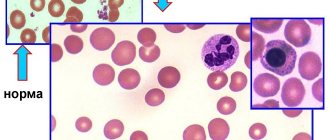
Anemia in AIHA with NTA is normochromic and slightly macrocytic in nature (due to the increased content of reticulocytes, which are larger in size than normocytes). When viewing a blood smear, noticeable polychromasia and anisocytosis, the presence of microspherocytes and normoblasts are noted (especially in patients with active hemolysis). The phenomenon of autoagglutination is rarely observed. The content of reticulocytes is increased. The lifespan of red blood cells is shortened.
Osmotic resistance is usually reduced due to changes in membrane properties by fixed autoantibodies, which also explains erythrocyte spherocytosis.
The number of leukocytes is most often slightly increased during hemolytic crises. The platelet count is normal or slightly reduced. In the presence, in addition to anti-erythrocyte antibodies, also platelet antibodies (Fisher-Evens syndrome), concomitant deep thrombocytopenia develops, causing severe hemorrhagic complications.
In the bone marrow, pronounced hyperplasia of the red line of hematopoiesis is detected, sometimes with megaloblastic features, due to a relative deficiency of folate, the consumption of which by actively proliferating normoblasts is significantly increased.
Hyperbilirubinemia is determined in plasma due to the unconjugated (indirect) fraction; the amount of stercobilin increases in feces, and urobilin in urine.
Diagnosis and differential diagnosis
The diagnosis of AIHA with NTA is established based on the detection of clinical and laboratory signs of acquired intracellular hemolysis and positive results of the direct antiglobulin Coombs test.
The Coombs test detects antibodies fixed on erythrocytes using antiglobulin antiserum obtained by immunizing animals with the globulin fraction of human plasma. Animal antiglobulin antiserum contains antibodies (such as complete agglutinins) directed against human globulins (Fig. 11). Since autoantibodies in AIHA with NTA are fixed on the surface of erythrocytes in their structure - globulins, under the influence of antiglobulin serum in a saline environment, agglutination of sensitized erythrocytes occurs, which confirms the presence of autoantibodies on their surface and the diagnosis of AIHA with NTA.
A negative direct Coombs test, however, does not exclude the presence of AIHA with NTA, but may also indicate a low density of autoantibodies on the surface of the erythrocyte (less than 200 molecules). In these cases, more sensitive tests are used to confirm the diagnosis: trypsin or papain-Coombs test and aggregate-hemmaglutination test, the resolution of which is more than 100 times higher than that of the standard direct Coombs test.
It is sometimes necessary to differentiate AIHA from NTA with drug-induced immune hemolytic anemia (LIHA), in which the direct Coombs test can also be positive. LIGA is based on a change in the antigenic structure of the erythrocyte membrane under the influence of a drug - a hapten, which results in the appearance of a new antigen on the erythrocyte and, as a result, the production of antibodies to it. Among the drugs that can cause immune hemolysis, mention should be made of α-methyldopa, cephalosporins, rifampicin, analgin, quinidine, paracetamol, etc. Therefore, in all patients with a positive direct Coombs test, when collecting anamnesis, it is necessary to exclude long-term use of medications, the withdrawal of which is usually leads to the cessation of hemolysis and makes the Coombs test negative.
Treatment
The main treatment method for AIHA with NTA is the prescription of corticosteroids (prednisolone or its analogues). On average, 1 mg of prednisolone is used per kg of patient weight (60-80 mg/day). At this dose, prednisolone is prescribed until hemoglobin levels normalize, jaundice and other laboratory signs of hemolysis disappear, which is sometimes accompanied by a negative direct Coombs test result. Then the dose of prednisolone is gradually reduced by 0.5-1 tablet. in 2-3 days until complete cancellation. Treatment with prednisolone leads to normalization of hemoglobin concentration in 75% of patients, although the direct Coombs test may remain positive.
If corticosteroids are ineffective, which becomes clear within 2 weeks of monitoring the patient, or if hemolysis relapses after its discontinuation, splenectomy is indicated, which can lead to a complete recovery of the patient or will increase the intervals between courses of corticosteroid therapy. For the treatment of patients resistant to prednisolone and in the absence of effect from splenectomy, cytostatics, immunosuppressants are used: imuran or 6-mercaptopurine at a dose of 100-150 mg per day, cyclophosphamide at a dose of 200 mg/day, chlorbutin 5-10 mg/day, cyclosporine 5 mg/kg/day and etc. In most cases, immunosuppressive therapy leads to an improvement in hematological parameters, however, stable remission is rarely observed.
In recent years, monoclonal antibodies to CD20 have been used in the treatment of resistant forms of AIHA - Rituximab (MabThera) intravenously 375 mg/m2 once a week for 4 weeks, the remission rate reaches 55-80%, maintenance therapy is usually not required. In severe cases, plasmapheresis may be used.
Intravenous administration of large doses of immunoglobulins, which neutralize autoantibodies and their effect on red blood cells, sometimes has a positive effect. Unfortunately, the abolition of intravenous immunoglobulin administration is accompanied by a relapse of the disease.
In cases of deep anemia with symptoms of hypoxia, only specially selected red blood cells can be transfused into patients with AIHA with NTA. For this purpose, at blood transfusion stations or blood banks, red blood cells of donors (usually from 15-20 people) are incubated (each sample separately) in the patient’s plasma at 370C. After this, a direct Coombs test is performed on each sample of donor red blood cells. If donor red blood cells are agglutinated by antiglobulin antiserum, they cannot be used for transfusion, because the membrane of donor red blood cells contains the same antigens against which the antibodies in the patient’s plasma are directed. Only those donor red blood cells that, after incubation in the patient’s plasma, gave a negative direct Coombs test can be transfused.
Course and prognosis
AIHA with NTA is characterized by an undulating course with alternating clinical well-being and relapses of the disease. The duration of the disease ranges from several months to many years. In ¼ of patients, complete recovery occurs with a persistent transition from a positive Coombs test to a negative one.
In general, the prognosis for AIHA with NTA should be considered serious, especially in secondary forms complicating chronic lymphocytic leukemia and SLE.
AIHA with complete thermal hemolysins (TH)
AIHA caused solely by hemolysins is quite rare. Warm hemolysins are more often found in patients with AIHA with NTA, which significantly worsens the prognosis for this disease.
Clinic
AIHA with TG can occur both acutely and chronically. The icterus of the skin and mucous membranes is slightly expressed. During hemolysis, abdominal pain and increased body temperature are possible. During a hemolytic crisis, thrombosis of various locations may develop.
The liver and spleen are usually not enlarged. Since the destruction of red blood cells by thermal hemolysins occurs with the active participation of activated complement inside the vessels, clinical manifestations include the appearance of black urine due to the content of oxidized hemoglobin or hemoglobinuria. In case of chronic intravascular hemolysis in a patient with AIHA with TG, hemosiderin is also present in the urine.
Laboratory data
Normochromic normocytic anemia, reticulocytosis. In plasma, the level of free hemoglobin increases and the concentration of haptoglobin decreases. In the urine - hemoglobinuria and hemosiderinuria. The participation of complement in the processes of hemolysis in AIHA with TG leads to a decrease in complement activity, and complement components C3, C4, C9 can be detected in a fixed state on the surface of erythrocytes using anti-complementary antiserum.
Diagnostics
The Coombs test in patients with hemolysin-induced AIHA is usually negative. This disease is diagnosed by an autohemolysis test: the patient’s blood, taken with citrate, is placed in a thermostat at 370C. After 30–40 minutes, redness of the plasma occurs due to the destruction of red blood cells by thermal autohemolysins present in the plasma.
Treatment
In the treatment of AIHA with TG, they try to use corticosteroids and immunosuppressants. Sometimes there is a development of long-term remission, however, complete recovery is rarely observed.
Forecast
The prognosis of hemolysin-induced AIHA depends on the frequency and intensity of hemolysis episodes.
Autoimmune hemolytic anemias with complete cold agglutinins
AIHA with complete cold agglutinins (CCA), or cold agglutinin disease (CAD), occurs mainly in older people. Symptomatic forms of CAB are more common in patients with chronic lymphocytic leukemia, Waldenström's macroglobulinemia, malignant lymphomas and monoclonal gammopathy. In young people, CAB may appear after infectious mononucleosis or mycoplasma pneumonia. CAB accounts for about 10–20% of all cases of AIHA. Women are somewhat more likely to suffer from HAB.
Clinic
KHAB is characterized by a chronic course. The disease begins gradually: a characteristic feature of the disease is poor tolerance to cold, under the influence of which patients develop “acrocyanosis” in the form of blue and pale skin of the fingers, ears and tip of the nose. Changes in skin color may be accompanied by sensory disturbances and pain. These symptoms, characteristic of Raynaud's syndrome, are usually reversible: they disappear as soon as the patient gets into a warm room. However, with prolonged exposure to the cold, gangrene may develop in areas where the body is cooled. The activity of hemolysis in CAB is usually low, so there may be only slight icterus of the skin and mucous membranes; the liver and spleen are usually not enlarged. The destruction of agglutinated erythrocytes is carried out by macrophages, i.e. hemolysis is intracellular in nature.
Laboratory data
Anemia is usually normochromic and normocytic in nature, and the hemoglobin level drops sharply to 80 g/l. When viewing blood smears, one notices the pronounced spontaneous autoagglutination of red blood cells, which makes them difficult to count.
Diagnosis and differential diagnosis
The diagnosis of CAB is based on the ability of the patient's serum in cold conditions to cause agglutination of group 0 donor red blood cells. Antibody titer, i.e. the degree of dilution at which the ability of the patient's serum to agglutinate red blood cells is preserved ranges from 1:1000 to 1:1000000. The titer of agglutinins and the temperature optimum for their action largely determine the clinical picture of the disease. Cold agglutinins belong to IgM and are usually directed against type I erythrocyte membrane antigens.
It is often necessary to differentiate CAP from diseases accompanied by impaired microcirculation in the cold: cryoglobulinemia and Raynaud's syndrome of vascular origin, often complicating rheumatic diseases (rheumatoid arthritis).
Treatment
Splenectomy and corticosteroids are usually ineffective for CAB. The use of immunosuppressants (chlorbutin, cyclophosphamide) gives positive results. Although the need for blood transfusions in CAB occurs relatively rarely, it should be remembered that patients with this disease can only be transfused with red blood cells washed in saline solution that do not contain complement on their surface.
In most cases, simple measures such as avoiding contact with cold, warming the patient and bed rest have a good effect.
Course and prognosis
The course of CAB is relatively benign with periods of worsening in winter and almost complete disappearance of symptoms in summer. There is practically no complete recovery from CHA, and cases of death are extremely rare.
In symptomatic forms of CAB, the prognosis is mainly determined by the underlying disease.
Autoimmune hemolytic anemia with biphasic cold hemolysins or paroxysmal cold hemoglobinuria (PHH)
UGS is one of the most rare types of AIHA. UCG is characterized by episodes (paroxysms) of intravascular hemolysis and hemoglobinuria, which are provoked by cooling.
PCH is caused by IgG antibodies, usually directed against the P-antigen of red blood cells and which are fixed on them at low temperatures. Hemolysis occurs with the participation of complement at body temperature.
Donat and Landsteiner at the beginning of the 20th century identified a causal relationship between advanced syphilis, especially its congenital form, and UGS. It has now been established that the role of syphilis in the development of UCG is small, however, this disease can complicate the course of some acute viral infections (measles, rubella, infectious mononucleosis, etc.) or occur without an established cause (idiopathic form).
Clinic
PCH occurs in all age groups, but more often in children. Both sexes are affected with equal frequency. The most characteristic feature of UGS is the appearance of black urine after local or general hypothermia, especially for quite a long time.
The disease begins acutely: a few minutes or hours after hypothermia, muscle pain, abdominal pain, general weakness, vomiting and stunning chills appear with an increase in body temperature to febrile levels. During this attack or shortly after it, black urine is passed. Subsequently, yellowness of the skin and sclera appears. In erased forms, which are observed in ½ of patients, all these symptoms are much less pronounced.
Laboratory data
Anemia develops only during a hemolytic crisis. The severity of anemia and reticulocytosis depend on the intensity and frequency of paroxysms of hemolysis.
The concentration of free hemoglobin in the blood increases (during a crisis). There is hemoglobinuria in the urine, hence its black color.
Diagnostics
The presence of biphasic autoantibodies of the Donath-Landsteiner type can be determined by hemolysis at 37° in pre-cooled blood, which is manifested by redness of the plasma (Donath-Landsteiner test). It has been established that biphasic hemolysins, unlike cold agglutinins, are rarely present in the blood in high titres.
The Coombs test, if performed at a low temperature, will be positive, but under standard conditions the results of this test are negative.
Treatment
In symptomatic forms of HCG, as a rule, spontaneous recovery is observed as the underlying disease is cured. In the treatment of idiopathic forms, the most important role is played by preventive measures aimed at preventing hypothermia.
Corticosteroids and splenectomy are ineffective for UCG.
Autoimmune anemia: prevention and treatment
Autoimmune anemia is a disease that speaks volumes about serious problems with the immune system. Immunomodulators are designed to eliminate such problems, but such a drug for anemia must meet a number of criteria. Firstly, high efficiency, secondly, safety, thirdly, lack of reaction with other drugs.
Modern science is ready to present an immunomodulator that meets all these requirements. Many years of research into cells of the immune system have led to the discovery of an amazing and unique molecule. It is called the transfer factor or transfer factor. Its specificity lies in the fact that it serves as a kind of microchip on which information about the functioning of the immune system is collected. Such molecules are inherent in all vertebrates, and are identical for all. This versatility has made it possible to isolate transfer factors from egg yolks and bovine colostrum. The choice of the latter product is not accidental, because it contains the maximum concentration of information cells. Colostrum is also the well-known “elixir of health”, since it is the source of a huge amount of antibodies that the mother passes on to her baby. Transfer factor is an immunomodulator of natural origin, which consists of a concentrate of information molecules. How does it work? The principle of operation of the drug is simple, like everything ingenious. Once in the body, transfer factor scans the DNA chain for damage and malfunctions. Next, information about the correct and effective functioning of the immune system is released. That is, transfer factors, like an experienced coach, identify the student’s weak points and provide information on exactly how to act in a given situation. This process occurs exclusively at the information level, and therefore does not affect the functioning of the body’s systems. There is also no interaction with any medications, which means Transfer Factor is completely safe.
Summarizing all of the above, we can say that taking Transfer Factor will allow the immune system to learn to work as efficiently as possible; distinguish between situations when it is necessary to strengthen the immune response, and when, on the contrary, to weaken it; allocate resources to protect the body that are most suitable for a given situation. This suggests that Transfer Factor is ideal for the treatment of autoimmune anemia, since it can correct errors in DNA and regulate the immune system properly.
Causes of autoimmune anemia
Autoimmune anemia, like any other autoimmune disease, is the result of a malfunction of the immune system. In turn, the reason for this failure can be a mutation of genes, damage to the DNA chain due to the influence of various harmful factors. And since DNA carries all the information necessary for the body to function, autoimmune anemia can also be inherited. For these reasons, successful treatment and prevention of autoimmune anemia lies primarily in optimizing the functioning of the immune system.