Porphyrias are a group of genetically determined pathologies that manifest themselves against the background of impaired synthesis of heme (an intermediate product of hemoglobin metabolism) and the accumulation of its toxic compounds in the human body. The symptoms of the disease are varied: patients may experience photosensitivity, skin rashes, chronic abdominal pain, partial or complete paralysis, and acute psychosis. Diagnosis of a metabolic disorder is carried out through molecular genetic tests and laboratory analyzes of biomaterials obtained by doctors from a child or adult. Drug therapy is aimed at reducing the concentration of toxic metabolites in the patient’s blood. Surgical interventions are performed in cases of complicated pathology.
general information
Porphyrin disease is diagnosed relatively rarely: Russian doctors detect no more than 12 cases per 100 thousand people. Various forms of pathology are becoming widespread in certain regions of the Earth. Thus, signs of porphyria cutanea tarda are often detected in residents of South Africa (1 case per 800 people). Acute intermittent type of disease is typical for residents of Northern Europe (1 case per 1000 people). Men and women are equally likely to suffer from various forms of heme synthesis.
List of sources
- Kuznetsova N.P., Pankov B.S., Chubarova A.S. and others. Porphyria. M., 1981. p. 66–14.
- Pustovoit Y.S., Pivnik A.V., Karpova I.V. Clinic, diagnosis and treatment of porphyria // Manual for doctors. Moscow, 2003
- Vorobyov A, Kravchenko S, Kremenetskaya A, Karpova I, Pustovoit Y. Acute intermittent porphyria: problems of diagnosis and treatment // Doctor. 2003 No. 2, pp. 8-13.
- Diagnosis and treatment of acute porphyrias: Clinical recommendations of the national hematological society / ed. Pustovoit Y.S., Kravchenko S.K., Shmakova R.G., Savchenko V.G. — 2021.
- Guide to hematology in 3 volumes / ed. Vorobyova A.I. – 2005 – T.3.
Reasons for development
The main factor provoking the development of the disease is genetic mutations in the carrier’s body. The only exception remains porphyria cutanea tarda - its origin is associated with liver pathologies or prolonged intoxication of the human body with salts of heavy metals.
Porphyrin disease is an inherited metabolic disorder. Individual symptoms of porphyria or attacks of accompanying psychosis can appear at any age. A specific set of signs of pathology is formed after the patient overcomes puberty.
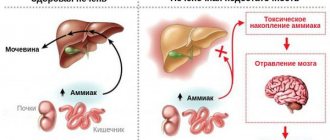
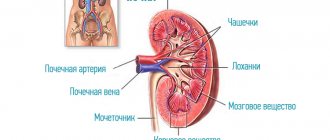
Heme synthesis consists of eight successive steps that occur under the action of enzymes. The production of complex protein compounds is encoded by a specific gene. Porphyrin disease leads to the formation of enzymatic defects. Different forms of the disease develop against the background of damage to various genetic codes. Heme consists of porphyrins combined with iron. The production of these compounds is carried out by the tissues of the liver and bone marrow. Hemoglobin is formed on the basis of heme. Violation of its synthesis leads to the formation of toxic metabolites that accumulate in the tissues of the human body.
The provoking factors for the manifestation of porphyrin disease are:
- excessive insolation;
- malnutrition and poor diet;
- systematic stress;
- excessive alcohol consumption;
- viral and bacterial infections;
- chronic intoxication with salts of heavy metals.
In some cases, exacerbation of pathology occurs due to patients taking antibiotics, anticonvulsants, non-steroidal anti-inflammatory drugs, and oral contraceptives.
ACUTE INTERMITTENT PORPHYRIA. CHANCE FOR LIFE.
Porphyrias are a heterogeneous group of metabolic diseases caused by a defect in one of the eight enzymes involved in heme synthesis. The history of the disease is associated with the crowned heads of England and myths about ghouls, who in reality most likely suffered from porphyria.
Porphyrias are rare diseases and occur with a frequency of 0.5–10 cases per 100,000 population. Currently in Russia, porphyria is included in the “List of life-threatening and chronic progressive rare (orphan) diseases leading to a reduction in the life expectancy of citizens or their disability,” approved by Government Resolution No. 403. At the moment, only a few hundred patients with porphyia have been diagnosed in Russia. According to statistics, for example, in the USA, more than 10,000 such patients are observed. Obviously, the small number of patients in Russia, compared to other countries, is associated with an insufficient level of awareness among doctors and a low level of laboratory diagnostics.
Most porphyrias are congenital diseases, predominantly with an autosomal dominant mode of inheritance. In some cases, metabolic disorders can be acquired and occur under the influence of factors that contribute to the inhibition of heme synthesis enzymes. The manifestations of various forms of the disease are determined by the nature of the accumulation and excretion of toxic heme precursors. The seven main forms of porphyria are divided into erythropoietic (erythropoietic uroporphyria and erythropoietic protoporphyria) and hepatic (the other five forms), depending on in which tissues porphyrins and their precursors accumulate. Depending on the clinical manifestations, all porphyrias can be divided into forms that occur predominantly with autonomic disorders or skin symptoms, and mixed forms. Porphyrias characterized by attacks with autonomic disorders are called hepatic, or acute. Four forms of hepatic porphyria are hereditary and one is acquired.
Attacks of Acute intermittent porphyria (AIP) are the most severe and are characterized by attacks of excruciating abdominal pain that mimic acute abdomen. The appearance of pain is often accompanied by nausea, vomiting, mental and neurological disorders. Many patients experience pain in the back and proximal extremities in combination with autonomic disorders, including arterial hypertension, orthostatic arterial hypotension, tachycardia and constipation.
For all types of acute porphyrias. during attacks. urine acquires a characteristic red, pink, or brown color (reddening of urine is possible when standing in the light in a glass jar), which is associated with an excess amount of porphobilin.
In general, acute porphyrias often manifest as attacks in the form of a combination of the following clinical symptoms: • Acute abdominal pain without peritoneal symptoms; • Red or pink urine (pigmenturia); • Autonomic disorders in the form of tachycardia, arterial hypertension, constipation, vomiting, dysfunction of the urethral sphincter; • Muscle weakness (usually peripheral paresis); • Mental disorders; • Epileptiform seizures; • Hypothalamic dysfunction in the form of central fever, hyponatremia.
Due to the variety of clinical manifestations and similarities with other, more common diseases, porphyria is often called the minor “pretender”. There are frequent cases of incorrect, erroneous diagnosis or cases of late diagnosis. There are cases when patients, due to an incorrect diagnosis, underwent unnecessary surgical intervention on the abdominal organs or were hospitalized for a long period in a psychiatric clinic.
In most patients, attacks occur at the age of 30–35 years, and in women 4–5 times more often than in men. In most cases, attacks develop under the influence of provoking factors, for example, the use of certain drugs, changes in hormonal levels (during the menstrual cycle or as a result of hormone replacement therapy), local or general anesthesia, sedative therapy, especially barbiturates, alcohol abuse, drug use ( amphetamines, cocaine or its derivatives), prolonged fasting, low-calorie diets, emotional or physical stress, acute viral and bacterial infections. Some drugs, due to their possible effect on heme metabolism, can provoke attacks of porphyria. Complete lists of such drugs are constantly updated and are provided on websites such as www.drugs-porphyria.org and www.porphyria-europe.com.
Clinical manifestations of hepatic porphyrias are not specific enough to diagnose the disease or determine its form. Therefore, for accurate diagnosis and proper treatment of porphyrias, it is necessary to conduct laboratory tests as early as possible (before the end of the attack), including identifying heme precursors in plasma, urine and feces and determining their levels. During attacks of porphyria, the levels of ALA and PBG in the urine always increase. If an attack of acute porphyria is suspected, it is necessary to perform a qualitative reaction with Ehrlich's reagent (an increase in the level of porphobilinogen (PBG) in urine more than 5 times higher than normal meets the criteria for an attack of acute porphyria) and quantify the level of each porphyrin in daily urine. Between attacks, urinary levels of porphyrin precursors (especially PBG and ALA) may normalize, making diagnosis more difficult during this period. The development of the technical basis for genetic research has made it possible to quickly conduct DNA analysis and identify carriers of mutations. These studies are used to confirm the diagnosis of porphyria attacks, assess the clinical significance of mutations, and identify asymptomatic carriers among the patient's relatives.
The mortality rate during an attack is unfortunately quite high at this time and is usually associated with respiratory failure and cardiac arrhythmias.
Treatment of attacks of acute porphyrias should be as early as possible and includes the administration of glucose and the drug hemin, leading to a decrease in the rate of ALA formation with subsequent normalization of clinical and biochemical parameters, including the level of ALA and PBG in the urine. For mild attacks, intravenous administration of glucose is indicated, followed by obligatory administration of hemin (under careful monitoring of sodium balance!). In severe attacks, treatment with hemin should be started as early as possible.
Porphyria is a serious and difficult to diagnose disease. However, a timely diagnosis and correct and timely treatment can reduce the degree of disability of patients and significantly reduce the risk of adverse outcomes.
The Russian expert group of doctors on porphyria, leading the all-Russian register of patients with porphyria, operates on the basis of the Federal State Budgetary Institution “Hematological Research Center” of the Ministry of Health of the Russian Federation (www.blood.ru). Yaroslav Sergeevich Pustovoit provides advisory support to doctors and patients with porphyria, observation and treatment of patients at the Hematology Research Center. (e-mail)
Providing Russian patients with porphyria with medications registered in the prescribed manner on the territory of the Russian Federation, on the basis of Federal Law No. 323 “On the fundamentals of protecting the health of citizens in the Russian Federation,” is carried out at the expense of the budgets of the constituent entities of the Russian Federation. In case of refusal to provide the necessary treatment, advisory and/or legal support for patients and doctors will be provided upon receipt of a written request on the website of the public organization of disabled people “Union of Patients and Patient Organizations for Rare Diseases” www.spiporz.ru/porfiriya
Additional information for doctors, patients and healthcare administrators in English is available on the European Porphyria Society website www.porphyria-europe.com
Official VKontakte group https://vk.com/porphiria
Etiology
Violation of the chemical composition of the enzymes involved in the production of heme leads to the formation of metabolites that are toxic to humans. Chronic forms of porphyria are characterized by the accumulation of protoporphyrin, coproporphyrin and uroporphyrin in tissues. Acute types of the disease develop against the background of increased concentrations of porphobilinogen and delta-aminolevulinic acid (DALA) in the blood.
The accumulation of porphyrins in the skin of children and adults occurs under the influence of ultraviolet radiation. A side effect of this process is lipid oxidation. Skin cells die when exposed to sunlight. Coproporphyrin provokes increased pigmentation of the dermis and increased hair growth. Deposition of protoporphyrin in the liver causes blockage of the bile ducts. Uroporphyrins lead to accelerated destruction of red blood cells in the spleen. DALK and porphobilinogen accumulate in nerve tissues and trigger the process of axonal degeneration.
Diagnosis of porphyria
The relative rarity of the pathology and the diversity of clinical manifestations cause regular diagnostic errors. This is especially true for the diagnosis of hepatic porphyrias. Often such patients are treated by a dermatologist and other specialists to no avail.
To diagnose the disease, it is necessary to conduct an analysis to determine the level of various fractions of porphyrins in the blood, urine and feces. Depending on which porphyrin fractions the level is elevated and what the ratio between different fractions is, one or another form of porphyria is diagnosed.
Types of pathology
Doctors use several typologies of porphyrin disease, based on clinical symptoms, localization of porphyrin metabolic disorders, or tissues involved. Most often, hematologists distinguish three forms of pathology: erythropoietic, acute hepatic and chronic hepatic.
The erythropoietic type of the disease is characterized by damage to the skin under the influence of sunlight. This form of pathology is considered by doctors as the most severe.
Acute hepatic types of porphyrin disease are characterized by paroxysmal course. The main target of toxic hemoglobin metabolites is the nervous system. In rare cases, the disease is complicated by photosensitivity.
Chronic liver diseases are often accompanied by the appearance of porphyria cutanea tarda. PKD may be hereditary or acquired pathology. Doctors consider it as the most favorable type of porphyrin disease for treatment.
Symptoms
Early symptoms of porphyria are nonspecific. The timing of the onset of the forms of the disease discussed above is different: the erythropic type manifests itself at 3–5 years, the acute hepatic type at 14–16 years, and the chronic hepatic type after 40 years. Following the manifestation of the pathology, patients are faced with specific symptoms.
Acute symptoms include abdominal pain, stool retention, a sharp increase in heart rate, increased blood pressure, and changes in urine color. The patient suffers from decreased sensitivity of the limbs, a feeling of aching in the bones and joints. Muscle weakness appears, and in severe cases, paralysis.
Later, seizures, delusions and hallucinations develop.
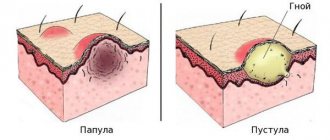
PKD is characterized by the formation of areas of hyperpigmentation of the skin under the influence of sunlight. Sallow or bronze-colored rashes appear on the face, neck, ears, and hands. With a complicated course of the pathology, the patient is faced with hypertrichosis: excess hair growth is localized in the frontotemporal region of the face. Blisters-pustules with liquid contents appear on the skin. The opened lesion forms erosion, after healing of which an atrophic scar is formed.
The erythropoietic form of porphyrin disease provokes the development in the patient of more pronounced signs of photosensitivity in comparison with PKP. A child or adult experiences severe pain when the skin is exposed to direct sunlight. Foci of erosion occupy a significant area and lead to the formation of rough scars that change a person’s appearance. Scar tissue reduces the mobility of the elbow and knee joints. The patient's urine takes on a red or pink tint, and the tooth enamel becomes reddish-brown. The skin of the face and the back of the hands becomes coarser and thickens.
Diagnostic measures
Diagnosis of porphyria is performed by a hematologist. The doctor examines the child or adult and includes in the history all the symptoms that may indicate disturbances in the process of heme synthesis. The patient will have to answer questions about medications taken, diet, and previous infectious diseases. The doctor asks the girls about the stability of the menstrual cycle, pregnancies and abortions.
The next stage of diagnosis is laboratory tests. Patients are prescribed:
- general clinical and biochemical blood tests;
- Ehrlich test;
- study of enzyme concentrations in the blood;
- PCR tests for hepatitis;
- molecular genetic research.
If there are appropriate indications, the child or adult attends a consultation with a dermatologist, nephrologist and gastroenterologist. Differential diagnosis allows doctors to exclude neurological and psychiatric pathologies from the patient’s medical history.
Prevention
There is no specific prevention. In order to minimize attacks, it is recommended:
- Avoid/minimize exposure to porphyrinogenic factors (regular lifestyle, avoid stress, taking potentially dangerous medications, drinking alcoholic beverages, fasting/strict diets, sun exposure).
- Patients over 50 years of age should undergo twice-yearly examinations to evaluate liver health.
- Rational employment.
- Carrying out preventive treatment/clinical examination of patients.
- If there is a porphyria patient in the family, it is necessary to carry out DNA diagnostics (detection of genetic mutations) and determine the activity of various enzymes of the heme synthesis cycle.
Therapeutic course
Treatment of acute and erythropoietic porphyria requires hospitalization of the patient. Individuals with PEP may receive therapy in an outpatient setting. Modern medicine does not have the means to completely eliminate disturbances in porphyrin metabolism. The efforts of doctors are aimed at eliminating the symptoms of the disease and the factors that provoke its exacerbation.
In acute forms of the pathology, a child or adult is prescribed adenosine triphosphate derivatives, which suppress the synthesis of toxic porphobilinogen. Further treatment involves the patient taking large doses of glucose and switching to a high-carbohydrate diet.
The erythropoietic type of the disease is almost untreatable. The main therapeutic measure is to limit the patient's exposure to sunlight. Erosions should be treated with antiseptic agents to prevent infectious lesions of the skin. Severe hemolysis (resolution of red blood cells) becomes an indication for removal of the spleen. In some cases, patients are indicated for bone marrow transplantation.
For porphyria cutanea tarda, men and women are prescribed plasmapheresis and drug therapy. The drugs used are designed to reduce the level of absorption of porphyrins in the gastrointestinal tract.
Treatment of porphyria
Porphyria is treated by a hematologist. Since the disease is genetic in nature, therapy is limited to reducing symptoms.
Causes and symptoms of porphyria
With erythropoietic porphyrias, sun protection comes to the fore: maximally closed clothing, wide-brimmed hats, sunscreens. To increase photoprotection, beta-carotene and nicotinic acid preparations may be recommended.
To delay the development of cirrhosis in hepatic porphyrias, patients are prescribed hepatoprotectors - for example, ursodeoxycholic acid preparations (Ursosan), which increase the resistance of hepatocytes (liver cells) to external toxic influences and thus reduce the activity of apoptosis (cell death).
To reduce the level of porphyrins in the body, bloodletting and antimalarial drugs may be recommended. Vitamins, ATP and cocarboxylase are prescribed as maintenance therapy.
Forecast
Porphyrin disease is a pathology with a poor prognosis. Patients with the erythropoietic type of disorder live 25–30 years after diagnosis and initiation of treatment. The cause of death in 85% of cases is secondary infections that develop against the background of weakened human immunity. Acute porphyrias are fatal in 15–20% of clinically recorded cases. The patient's death occurs as a result of paralysis of the respiratory muscles.
PKP remains a relatively benign form of porphyrin disease. Men and women are advised to avoid factors that provoke complications: poor diet, stress, excessive insolation.