Klinefelter syndrome is a genetic disease characterized by an additional female sex chromosome X (one or even several) in the male karyotype XY . At the same time, insufficient sex hormones are produced in the male gonads - the testicles.
As you know, the human genetic set has 46 chromosomes, of which 22 pairs are called somatic, and the 23rd pair is called sexual.
Women have a pair of sex chromosomes XX , and men have XY . Klinefelter syndrome requires the presence of a male Y chromosome, so despite the additional X chromosomes, patients are always male.
Classification: types of karyotypes in Klinefelter syndrome
Based on the number of additional X chromosomes, the following variants of Klinefelter syndrome are distinguished:
- 47,XXY - the most common
- 48,ХХХY
- 49,XXXXY
In addition, Klinefelter syndrome also includes male karyotypes that include, in addition to additional X chromosomes, an additional Y chromosome - 48,XXYY . And finally, among patients with this syndrome there are individuals with a mosaic karyotype 46,XY / 47,XXY (that is, some of the cells have a normal chromosome set).
History of the discovery of the syndrome
The syndrome got its name in honor of Harry Klinefelter, a doctor who first described the clinical picture of the disease in 1942. Klinefelter and colleagues published a study of 9 men with common symptoms such as low body hair, eunuchoid body type, tall stature, and small testicles. Later, in 1956, geneticists Plunkett and Barr (E.R. Plunkett, M.L. Barr) discovered sex chromatin bodies in the nuclei of cells of the oral mucosa in men with Klinefelter syndrome, and in 1959 Polanyi and Ford (P.E. Polanyi, SE Ford) and colleagues showed that patients have an extra X chromosome in their chromosome set.
Active research into this pathology was conducted in the 70s in the USA. Then all newborn boys were subjected to karyotyping, as a result of which it was possible to reliably identify the prevalence and genetic characteristics of Klinefelter syndrome.
Interestingly, mice can also have XXY sex chromosome trisomy, making them useful models for studying Klinefelter syndrome.
Prevalence of the disease
Klinefelter syndrome is one of the most common genetic diseases: for every 500 newborn boys, there is 1 child with this pathology.
In addition, Klinefelter syndrome is the third most common endocrine pathology in men (after diabetes mellitus and thyroid pathology) and the most common cause of congenital reproductive dysfunction in men.
To date, about half of cases of Klinefelter syndrome remain unrecognized. Often such patients seek help for infertility, erectile dysfunction, gynecomastia, osteoporosis, anemia, etc. without a previously established diagnosis.
Etiology and causes of the disorder
Klinefelter syndrome is a genetic disease that is not inherited because patients, with rare exceptions, are infertile. Pathology, as a rule, occurs as a result of a violation of chromosome divergence in the early stages of the formation of eggs and sperm. At the same time, Klinefelter syndrome, which occurs due to a disorder in female reproductive cells, occurs three times more often. Mosaic forms are caused by pathology of cell division in the early stages of embryogenesis, therefore some of the cells in such patients have a normal karyotype. The reasons for nondisjunction of sex chromosomes and disruption of cell division at the earliest stages of embryogenesis are still poorly understood. Unlike other chromosomal diseases, the effect of parental age is absent or only slightly expressed.
Forecast
If the manifestations of Klinefelter syndrome are mild, there are no changes in the intellectual sphere, and there are no serious consequences, then the prognosis is favorable. With timely treatment and correction of possible complications, patients can live a full life.
If Klinefelter's symptoms are more pronounced and there are mental disorders and serious chronic conditions, then the prognosis noticeably worsens. In such patients, treatment should be comprehensive, including not only medication, but psychocorrection and rehabilitation.
Severe concomitant diseases of internal organs lead to early mortality and disability.
Lack of testosterone leads to spontaneous fractures and acute heart problems, such as myocardial infarction.
Early signs
Unlike most diseases associated with a violation of the number of chromosomes, the intrauterine development of children with Klinefelter syndrome proceeds normally, and there is no tendency to premature termination of pregnancy. So in infancy and early childhood it is almost impossible to suspect pathology. Moreover, clinical signs of classic Klinefelter syndrome usually appear only in adolescence. However, there are symptoms that suggest the presence of Klinefelter syndrome in the prepubertal period:
- high growth (peak height increase occurs between 5–8 years);
- long legs (disproportionate physique);
- high waist.
Some patients experience some delay in speech development.
In adolescence, the syndrome often manifests itself as gynecomastia, which with this pathology has the appearance of bilateral symmetrical painless enlargement of the mammary glands. Since this type of gynecomastia is often observed in completely healthy adolescents, this symptom often goes unnoticed. Normally, teenage gynecomastia disappears without a trace within several years, but in patients with Klinefelter syndrome, reverse involution of the mammary glands does not occur. In some cases, gynecomastia may not develop at all, and then the pathology manifests itself as signs of androgen deficiency already in the postpubertal period.
Sex chromosomes: numerical and structural abnormalities
Table of contents
- Introduction
- Summary
- Abnormalities in chromosome number (aneuploidy)
- Monosomy on the X chromosome (45.X, or Shereshevsky-Turner syndrome)
- 47.XXY Klinefelter syndrome
- 47.XYY
- 47.XXX
- Other diseases
- Mosaicism 45,X/46,XX
- Mosaicism 45,X/46,XY
- Structural abnormalities of chromosomes
- Isochromosome Xq
- Xp22.11 deletion
- Xp22.3 deletion
- Xp22 SHOX deletions
- Xp11.22 deletions
- Xp.22.31 duplications
- ME2CP duplication syndrome
Introduction
Pathologies of sex chromosomes are associated with a violation of their number (i.e., aneuploidy, for example, monosomy of the X chromosome) or with structural defects (for example, genomic rearrangements such as MECP2
). The frequency of congenital chromosomal mutations is at least 1:400.
Summary
Pathologies of sex chromosomes can be caused by a violation of their number (aneuploidy) or structural defects.
The most common sex chromosome aneuploidies are: 45.X (Turner syndrome); 47,XXY (Klinefelter syndrome); 47,XYY; and 47,XXX. Mosaicism of sex chromosomes with the presence in the body of cells with a normal genotype is not uncommon. The two most common types of sex chromosome mosaicism are 45,X/46,XX and 45,X/46,XY. The severity of phenotypic manifestations in patients with mosaicism corresponds to the proportion of abnormal cells.
Structural pathologies of the X and Y chromosomes primarily include isochromosomes, deletions, duplications, ring chromosomes, and translocations.
One example of a genomic disorder is MECP2
in men, expressed in the presence of muscle hypotonia, severe mental retardation, delayed speech development, swallowing disorders, frequent respiratory infections, as well as convulsive seizures (tonic-clonic seizures that cannot be treated).
Abnormalities in chromosome number (aneuploidy)
The most common sex chromosome aneuploidies are 45.X (Shereshevsky-Turner syndrome); 47,XXY (Klinefelter syndrome); 47,XYY and 47,XXX with an incidence of approximately 1/2500, 1/500 to 1/1000, 1/900 to 1500, and 1/1000, respectively. Mosaicism of sex chromosomes with the presence in the body of cells with a normal genotype is not uncommon. The two most common types of sex chromosome mosaicism are 45,X/46,XX and 45,X/46,XY. The severity of phenotypic manifestations in patients with mosaicism corresponds to the percentage of abnormal cells.
Monosomy on the X chromosome (45.X, or Shereshevsky-Turner syndrome)
Most patients with Shereshevsky-Turner syndrome have monosomy on the X chromosome, karyotype 45.X. Other forms of the syndrome include mosaicism on the X chromosome, such as 45,X/46,XX or 45,X/46,XY with partial deletion of the Y chromosome. Some patients have a structural abnormality of the second X chromosome (eg, long arm isochromosomy of the X chromosome or deletion of the short arm). Deletions involving the distal part of the short arm of the Y chromosome are also associated with the Turner syndrome phenotype, since in this case the patients lack the so-called anti-Turner genes (SHOX, RPSY4 and ZFY). Deletions of the short arm of the X chromosome have also been associated with the Turner syndrome phenotype. Most are isolated cases.
Shereshevsky-Turner syndrome is characterized by short stature and some of the following: facial dysmorphia including low-set ears, skin folds on the neck, shield-shaped chest (wide, with a large distance between the nipples), lymphedema, valgus deformity of the elbow joint, short fourth metacarpal bone, hypoplasia of the nail plates, age spots and congenital heart defects. Among heart defects, the typical and most common are vascular defects and coarctation of the aorta. In addition, patients suffering from Turner syndrome develop streak-shaped gonads, ovulation disorders and delayed sexual development. Defects in kidney development (horseshoe kidney) also occur. Lymphedema of the lower extremities may be the only clinical sign observed in newborns. Individuals with Turner syndrome who carry genetic material from the Y chromosome have an increased risk of developing gonadoblastoma.
47.XXY Klinefelter syndrome
Klinefelter syndrome is the most common pathology of the number of sex chromosomes causing primary hypogonadism. The 47,XXY karyotype is the result of sex chromosome nondisjunction and can be either maternal or paternal in origin. Most cases of the disease are detected postnatally and are diagnosed when determining the causes of infertility, identifying gynecomastia, cryptorchidism, or neurological disorders.
Rice. Nondisjunction of sex chromosomes
Newborn boys with a 47,XXY karyotype are phenotypically normal, with physiologically normal male external genitalia and without any visible dysmorphia. The main clinical manifestations of Klinefelter syndrome, including tall stature, small testicles and infertility (azoospermia), become pronounced in the postpubertal period. Patients with Klinefelter syndrome have an increased risk of mental disorders, autism spectrum disorders, and social problems. Patients diagnosed with Klinefelter syndrome should have their neurological status assessed and referred to an endocrinologist.
47.XYY
Individuals with a 47,XYY karyotype are tall and may experience moderate delays in motor and speech development. Many of them require increased attention to learning, but, as a rule, they all study in basic comprehensive schools. Pubertal development is normal and most boys are fertile. Because the phenotype is mild and there are no associated health problems, many individuals with a 47,XYY karyotype remain undiagnosed throughout their lives.
It has previously been reported that men with 47.XYY have increased aggression, which is reflected in their aggressive behavior. However, subsequent large-scale joint studies of European and American geneticists showed that the statistics of increased criminal activity of men with XYY correlated with their low socio-economic status due to low IQ (about 10 points), which led to certain difficulties with the law and, more often, minor offenses. Individuals with 47,XYY have higher rates of attention deficit hyperactivity disorder and autism spectrum disorder. Neurodevelopmental assessment is recommended for these patients, given the high prevalence of learning difficulties and behavioral problems.
47.XXX
47,XXX (also known as trisomy X) is the most common pathology of the sex chromosomes in women. Trisomy X is diagnosed in utero during genetic screening. Women with a 47.XXX karyotype do not have an increased risk of developing a fetus with chromosomal abnormalities.
A study of 155 women with a karyotype of 47.XXX showed that 62 percent of them were physically normal. Thus, most individuals with a 47.XXX karyotype are never diagnosed. Women with 47.XXX have high growth; (average head circumference varies between the 25th and 35th percentile, but by adolescence for many it can reach the 80th percentile). Sexual maturity and fertility are most often normal, but premature ovarian failure may occur.
A subsequent study of eleven infants with a karyotype of 47.XXX showed that the girls' IQ from birth was 15-20 points lower than that of their brothers. Therefore, it is recommended to monitor developmental delays and identify the presence of psychological problems in the future.
Other diseases
More than one hundred cases of the 49,XXXXY karyotype have been reported, with at least twenty cases of 49,XXXXX and a few cases of 49,XYYYY. There is a direct relationship between the number of additional sex chromosomes and the severity of phenotypic manifestations in patients. A study of tetra- and pentasomy of sex chromosomes concluded that polysomy on the X chromosome is associated with more severe consequences than polysomy on the Y chromosome. It has been shown that IQ levels decrease by 10 points for each extra X chromosome from the normal number.
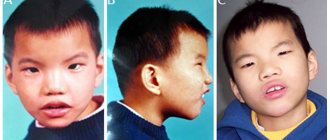
49,XXXXY Characteristic clinical features of the XXXXY karyotype are a sunken bridge of the nose with a wide or raised nasal tip, widely spaced eyes, eyelid-nasal folds, skeletal pathologies (especially radioulnar synostosis), congenital heart diseases, endocrine disorders, and a high degree of hypogonadism and hypogenitalism. Severe mental retardation and moderate short stature are also common. Although individuals with this karyotype are often classified as cases of Klinefelter syndrome, all the characteristic features of XXXXY point quite clearly to this phenotype.
49,XXXXX Women with karyotype 49,XXXXX (pentasomy on the X chromosome) always have mental retardation. Other manifestations such as craniofacial, cardiovascular and skeletal pathologies are quite variable. Patients with pentasomy X may exhibit features similar to those seen in Down syndrome. Radioulnar synostosis is also common in patients with a large number of X chromosomes. Some patients have 48,XXXX and 49,XXXXX mosaicism.
Mosaicism 45,X/46,XX
This is the most common sex chromosome mosaicism and is diagnosed by amniocentesis and prenatal karyotyping. Individuals with this type of mosaicism have milder clinical features of Turner syndrome. Many women went through puberty and were able to reproduce.
Of 156 prenatally diagnosed cases of 45.X/46.XX mosaicism, 14% of cases had an abnormal outcome. There were two stillbirths and 20 cases of abnormal phenotype (12 had some features of Turner syndrome, and the remaining 8 were abnormal, possibly unrelated). More than 85% of girls had a normal phenotype at birth, or it was established as a result of medical termination of pregnancy. However, the main features of Turner syndrome (such as short stature and absence of secondary sexual characteristics) appear only in childhood or adolescence, and are not noticed in infancy. In some women with a normal phenotype, with impaired ovarian function, 45,X/46,XX mosaicism is detected.
Mosaicism 45,X/46,XY
Mosaicism with the presence of 45,X/46,XY has a wide phenotypic spectrum. For example, in a retrospective series of 151 postnatally diagnosed cases of 45,X/46,XY mosaicism, 42% of patients were female in phenotype, with typical or atypical Turner syndrome present. Another 42% had indeterminate external genitalia and asymmetrical gonads (mixed gonadal dysgenesis), and finally, 15% had a male phenotype with incomplete masculinization. Thus, all cases diagnosed postnatally were phenotypically abnormal. In contrast, among 80 prenatally diagnosed cases of 45,X/46,XY 74 mosaicism, 92.6% were phenotypically normal boys. This may explain the fact that children or adults with mosaicism but a normal phenotype would be unlikely to seek medical help (referral bias).
Structural abnormalities of chromosomes
Structural pathologies include primarily isochromosomes, deletions, duplications, ring chromosomes and translocations.
Isochromosome Xq
Isochromosome of the long arm of the X chromosome, isoXq or i(Xq), in which the short arm (p) is eliminated (absent/reduced) and replaced by an exact copy of the long arm (q), is the most common abnormality of the sex chromosomes.
The presence of structural pathology is not associated with an increased age-related risk in parents. Isochromosomy 46,X,i(Xq) can be an expression of mosaicism, when two genetically different cell populations are present in the body: normal - 46,XX and 45,X.
Isochromosomes Xq and Xy are associated with Turner syndrome, possibly because the major anti-Turner gene SHOX is located on the distal part of the short arms of the X and Y chromosomes (in the pseudoautosomal regions). Isochromosome Xq is also detected in patients with one of the variations of Klinefelter syndrome, 47,X,i(Xq),Y.
Xp22.11 deletion
The Xp22.11 deletion includes the PTCHD1
.
Identification was reported in several families with autistic disorders, as well as in three families with mental retardation. PTCHD1
gene is a candidate gene for X-linked mental retardation with or without autism. The function and role of this gene is unknown.
Xp22.3 deletion
Deletion of this region is often associated with microphthalmia and linear skin defects syndrome (MLS) and is an X-linked dominant disorder, that is, lethal in men and therefore seen only in women. The gene in this region encodes mitochondrial cytochrome c synthase ( HCCS
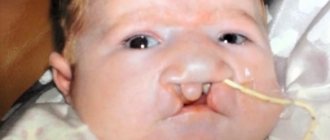
). The clinical manifestation of MLS is expressed by the presence of microphthalmia and anophthalmia (unilateral or bilateral) and linear skin defects, mainly of the face and neck, which resolve over time. Structural pathologies of the brain, developmental delays, and seizures (seizures) are also part of the clinical picture. Cardiac abnormalities (such as hypertensive cardiomyopathy and arrhythmia), short stature, hiatal hernia, nail dystrophy, preauricular fistula, hearing loss, genitourinary malformations (malformations, malformation) are also common clinical phenomena.
The screening assessment includes an ophthalmological and dermatological examination, assessment of general development, echocardiogram, brain magnetic resonance imaging (MRI) and electroencephalogram (EEG).
Xp22 SHOX deletions
The Xp22 deletion includes the SHOX gene, a mutation of which causes idiopathic short stature. The SHOX gene is located in pseudoautosomal region 1 of the X and Y chromosomes. This gene is believed to be responsible for short stature in Turner syndrome, and haploinsufficiency of this gene causes Lery-Weillian dyschondrosteosis. Lery-Weill's dyschondrosteosis is characterized by short stature, most pronounced in women, as well as chronic subluxation of the hand (deformation of the wrist bones, Madelung deformity). Homozygous deletions of the SHOX gene cause Langer's dysplasia, a more severe form of metaphyseal dysplasia. SHOX gene deletions are easily detected in patients with short stature, without any other specific features in their skeletal structure. More than 60% of SHOX rearrangements are gene deletions; in the absence of deletions, comparative genomic hybridization followed by sequencing to identify and establish point mutations is the clinical investigation of idiopathic short stature.
Xp11.22 deletions
Deletions of the Xp11.22 region include the PHF8 gene (encoding the finger protein PHD8), mutations of which have been associated with mental retardation, cleft lip/palate, and autistic disorders.
Mutations with deletion of the PHF8 gene are associated with the X-linked mental retardation syndrome, Siderius-Hamel syndrome.
Xp.22.31 duplications
Duplications at the Xp.22.31 locus are frequently described in the literature. There has been much debate about whether this duplication is pathogenetic or benign, given the difficulty of determining the consequences of gene copy number variation. This duplication affects the steroid sulfatase gene. The result is a genetic defect, a mutation in the steroid sulfatase gene, which is expressed in a decrease in its activity or the absence of its synthesis. Deletion of this gene is associated with X-linked ichthyosis in men. This duplication is observed in patients with mental retardation. However, it is detected both in healthy relatives of these patients and in the general population. Although duplications of this gene may not have phenotypic manifestations, triplications have been consistently associated with mental disorders. FISH diagnostics ultimately makes it possible to differentiate duplications from triplications (to recognize an increase in the copy number of a gene).
ME2CP duplication syndrome
Mutations in the gene encoding methyl-binding-CpG terminal protein 2 ( ME2CP
), located in Xq28, responsible for Rett syndrome.
Duplication of this region has little or no phenotypic significance in women, probably due to inactivation of the pathological X chromosome. Men with this mutation are greatly weakened. The presence of duplication is clinically expressed in the presence of severe muscle hypotonia, severe mental retardation, delayed speech development, swallowing disorders (difficulty eating), frequent respiratory infections and convulsive seizures, including tonic-clonic ones, which are sometimes untreatable. Many patients with this duplication were diagnosed with autism or a similar type of disorder. Similar to what is seen in Rett syndrome, patients with ME2CP
experience developmental regression.
In addition, they develop ataxia, and progressive muscle spasticity of the lower body often leads to loss of ambulation. Gastrointestinal problems and severe constipation were noted. Duplication often affects the interleukin receptor antagonist 1 ( IRAK1
) gene, which may play a role in the immune pathologies observed in this group of patients. The prognosis is poor, and most men with this duplication die before the age of 30 due to secondary respiratory infections. Triplication of this region results in an even more severe phenotype in men.
Screening examinations of these patients include an EEG, assessment of swallowing function, and assessment of humoral and cellular immunity. Treatment may include treatment for muscle hypotonia and spasticity, speech therapy (speech therapy), the use of a gastrostomy tube for feeding problems, and treatment for respiratory infections.
The translation of materials from the UpTodate website was prepared by specialists from the Center for Immunology and Reproduction.
Symptoms of androgen deficiency in Klinefelter syndrome
Androgen deficiency in Klinefelter syndrome is associated with gradual testicular atrophy, which leads to decreased testosterone synthesis. The degree of androgen deficiency varies dramatically.
First of all, the external signs of hypogonadism attract attention:
- scanty facial hair or its complete absence;
- female-pattern pubic hair growth;
- there is no hair on the chest or other parts of the body;
- small volume of the testicles (2–4 ml) and their dense consistency (pathognomonic sign).
Since degeneration of the gonads, as a rule, develops in the postpubertal period, in most patients the size of the male genital organs, with the exception of the testicles, corresponds to age norms.
Patients may complain of weakened libido and decreased potency. Many men with Klinefelter syndrome do not experience sexual desire at all, while some, on the contrary, start a family and live a normal sex life. The most constant sign of pathology is infertility; it is this that most often becomes the reason for such patients to consult a doctor. 10% of men with azoospemia have Klinefelter syndrome.
All patients with spermatogenesis disorders must have their karyotype determined to exclude or confirm the diagnosis of Klinefelter syndrome.
Androgen deficiency leads to the development of osteoporosis, anemia and skeletal muscle weakness. In a third of patients, varicose veins of the legs can be observed.
Androgens affect metabolism, so patients with Klinefelter syndrome are prone to obesity, impaired glucose tolerance and type 2 diabetes.
The predisposition of such patients to autoimmune diseases (rheumatoid arthritis, systemic lupus erythematosus, autoimmune thyroid diseases and others) has been proven.
Psychological characteristics
The IQ of patients with classic Klinefelter syndrome varies from below average to well above average. However, in all cases there is a disproportion between the general level of intelligence and verbal abilities, so that patients with a fairly high IQ often experience difficulties in perceiving large volumes of material by ear, as well as in constructing phrases containing complex grammatical structures. Such features cause patients a lot of trouble during the training period and often continue to affect their professional activities.
Data on the psychological characteristics of patients with Klinefelter syndrome are quite contradictory, but most experts assess patients as modest, timid people with somewhat low self-esteem and increased sensitivity. There is evidence that patients with Klinefelter syndrome are prone to homosexuality, alcoholism and drug addiction. It is difficult to say whether the mental characteristics of such patients are caused by the direct influence of a chromosomal abnormality, or whether it is a reaction to problems in the sexual sphere.
With regard to different cytogenetic variants of Klinefelter syndrome, the rule is true that with an increase in the number of additional X chromosomes, the number and severity of pathological symptoms increases.
Classification
Normally, a human karyotype contains 46 chromosomes. It distinguishes between the sex chromosomes X - chromosome (female) and Y - chromosome (male).
In healthy men, the karyotype has both chromosomes. In Klinefelter syndrome, the karyotype contains additional female sex chromosomes.
Extra female sex chromosomes have a great impact on the condition of the body with Klinefelter syndrome. The mutation suppresses testosterone levels, reducing its concentration, leading to pathologies of the reproductive system.
Chromosome combinations vary in patients with Klinefelter syndrome. The karyotype can be of several types:
- 47 XXY;
- 48 XXYY;
- 48 XXXY;
- 49 XXXYY;
- 49 XXXXYY;
- 47 XXY (mosaicism).
| Karyotype options | Peculiarities |
| 47 XXY | Like all forms of Klinefelter syndrome, they are accompanied by infertility, a “female” body structure, underdevelopment of the genital organs, and minor mental disorders. |
| 48 XXYY | Mental retardation is weakly expressed. Features of the body structure (long arms, disproportionate body type), underdevelopment of the penis, testicles, gynecomastia come to the fore. |
| 48 XXXY | Characterized by moderate changes in the mental sphere. Mental retardation is mild, there is a delay in speech and motor function, accompanied by aggression and low self-esteem. |
| 49 XXXYY | It manifests as moderate to severe mental retardation. Accompanied by malformations of the body. |
| 49 XXXXYY | One of the severe forms, accompanied by severe mental disorders and abnormal development of the genital organs. Mental retardation is pronounced. Various malformations of internal organs and concomitant diseases are observed. Manifested by severe testosterone deficiency. |
| 47 XY (mosaic shape) | Reproductive function is preserved, they can have children, spermatogenesis is not impaired. This variant of Klinefelter syndrome is asymptomatic; a person may not suspect that he has the disease for the rest of his life. |
Diagnosis of Klinefelter syndrome
In many countries, Klinefelter syndrome is often diagnosed before the birth of a child, since many women of late childbearing age, due to the high risk of genetic defects in future offspring, use prenatal genetic diagnosis of the fetus. Often, prenatal detection of Klinefelter syndrome is a reason for termination of pregnancy, including on the recommendation of doctors. In Russia, analysis of the karyotype of an unborn child is extremely rare.
If Klinefelter syndrome is suspected, a laboratory blood test is performed to determine the level of male sex hormones. Differential diagnosis with other diseases that occur with manifestations of androgen deficiency is necessary. An accurate diagnosis of Klinefelter syndrome is made based on studying the karyotype (set of chromosomes) of the patient.
Tests needed to confirm the diagnosis
| Analyzes | results |
| Karyotype | 47,ХХY (80% of cases) 48,ХХYY 48,ХХХY 49,ХХХY 46,ХY/47,ХХY |
| Concentration of LH, FSH | Increased, especially FSH |
| Total testosterone concentration | Most often reduced (in some cases normal due to an increase in sex steroid-binding globulin SSSG or at the initial stage of disease development) |
In all men with sharply increased concentrations of gonadotropins, it is necessary to exclude Klinefelter syndrome, since often the first laboratory sign of this genetic pathology is an increase in the concentration of gonadotropins in the blood with normal levels of total testosterone.
Klinefelter syndrome must be differentiated from other forms of primary hypogonadism. In any case, if the level of FSH in the blood increases, it is necessary to determine the karyotype to exclude, first of all, Klinefelter syndrome.
Prevention
There is no specific prevention for Klinefelter syndrome. There are only a few activities that reduce the risk of developing the disease.
Preventive actions:
- planning pregnancy before age 30;
- quitting smoking and alcohol during pregnancy;
- regular screening tests during pregnancy;
- genetic consultation for women aged 35 to 40 years.
If Klinefelter syndrome is detected during screening, the doctor may suggest termination of the pregnancy. But in this case, the decision remains with the woman.
Treatment
Treatment goals for Klinefelter syndrome:
- Restoring normal testosterone levels
- Restoration of sexual function
- Elimination of metabolic disorders
In case of clinically pronounced pathology, lifelong replacement therapy with testosterone preparations is necessary. Adequate therapy allows not only to improve the appearance and general well-being of the patient, but also to restore the ability to have a normal sexual life. In addition, replacement therapy prevents the development of osteoporosis and relieves muscle weakness. At a young age, treatment should begin immediately after diagnosis. For Klinefelter syndrome, it is better to use long-acting testosterone drugs:
- a mixture of testosterone esters in the form of an oil solution, injections of which must be done 2-3 times a month;
- testosterone undecanoate in the form of an oil solution - a depot preparation with a slow release of the active substance - injections once every 3 months.
Hormone treatment for the presence of the X chromosome in men should be permanent. The dose of the drug is selected individually under the control of testosterone and LH levels in the blood serum.
Already developed gynecomastia in Klinefelter syndrome does not undergo involution even with adequate treatment, so it is often necessary to resort to surgical correction (mastectomy).
To prevent concomitant diseases such as obesity and type 2 diabetes, patients are advised to adhere to a diet and monitor their own weight.
Patients with Klinefelter syndrome should be monitored at least once every 6–12 months. It should include the following studies:
- complete blood count to assess hemoglobin and hematocrit levels;
- hormonal blood test, including determination of testosterone and LH (carried out against the background of drug therapy 1–2 days before the next testosterone injection);
- densitometry (all patients who had osteopenia or osteoporosis at the time of diagnosis).
The introduction of intracytoplasmic sperm injection (ICSI) and data on the possibility of the presence of germ cells in the testes in patients with Klinefelter syndrome predetermined the use of artificial insemination for this category of patients, some attempts were successful.
Dispensary observation
Clinical observation is carried out throughout life by an endocrinologist and other specialists as necessary.
In childhood, observation is carried out by a pediatrician. In the first year of life, the frequency of examinations is 1 time per month (as in healthy children). Then the number is reduced to 1 time every 3 months.
As the child grows up, an endocrinologist joins the observation. Since such children develop endocrine problems.
In adults, if there are no complaints, examinations can be carried out once a year.
In addition, additional studies are prescribed:
- clinical blood test;
- blood biochemistry;
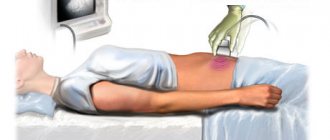
- Ultrasound of the abdominal organs;
- Ultrasound of the penis and testicles;
- general urine analysis;
- ECG or ultrasound of the heart.
Specialist consultations are carried out. And special attention is paid to consultations with a cardiologist, urologist, reproductive specialist, and psychotherapist.